

Abstract
A myopathy with severe exercise intolerance and myoglobinuria has been described in patients from northern Sweden, with associated deficiencies of succinate dehydrogenase and aconitase in skeletal muscle. We identified the gene for the iron-sulfur cluster scaffold protein ISCU as a candidate within a region of shared homozygosity among patients with this disease. We found a single mutation in ISCU that likely strengthens a weak splice acceptor site, with consequent exon retention. A marked reduction of ISCU mRNA and mitochondrial ISCU protein in patient muscle was associated with a decrease in the iron regulatory protein IRP1 and intracellular iron overload in skeletal muscle, consistent with a muscle-specific alteration of iron homeostasis in this disease. ISCU interacts with the Friedreich ataxia gene product frataxin in iron-sulfur cluster biosynthesis. Our results therefore extend the range of known human diseases that are caused by defects in iron-sulfur cluster biogenesis.
Introduction
Myopathy with deficiency of succinate dehydrogenase and aconitase was initially reported by Larsson et al. in 14 patients from five families in northern Sweden.1–3 The disease is characterized by lifelong severe exercise intolerance, in which minor exertion causes fatigue of active muscles, shortness of breath, and cardiac palpitations in association with lactic acidosis. Patients also experience episodes of rhabdomyolysis associated with muscle swelling and pain, weakness that may be profound, and myoglobinuria. Physiological investigations of these patients during exercise showed impaired muscle oxidative phosphorylation. Low maximal muscle oxygen extraction was associated with exaggerated circulatory responses, where the increase in cardiac output relative to oxygen utilization during exercise was four to six times normal. Biochemical studies indicated a deficiency in succinate dehydrogenase (SDH)4 and aconitase5 activities and the presence of electron-dense, iron-rich mitochondrial inclusions.5 A more generalized abnormality of muscle mitochondrial iron-sulfur-cluster-containing proteins, including the mature Rieske iron-sulfur protein of complex III and several subunits of complex I, was subsequently demonstrated,6 with the heart and vascular smooth muscles spared. In the current study, we found a common region of homozygosity in three patients from three families originating from northern Sweden and confirmed a founder haplotype. Within that interval, we further identified a single intronic mutation in the gene for the iron-sulfur cluster scaffold protein ISCU. This homozygous mutation strengthens a weak splice acceptor site and results in reduced levels of ISCU mRNA and protein, leading to adverse effects on iron-sulfur proteins and intracellular iron homeostasis.
Material and Methods
Affected Individuals and Families
We studied three affected individuals (P1, P2, and P3), aged 37, 39, and 66 years, respectively, from three different pedigrees and the unaffected offspring (H1), aged 44 years, of one of the affected individuals (P3). There was no known consanguinity in any of the three families, all of which originated in northern Sweden. Patient P1 has been extensively reported elsewhere,5,6 and patient P3 belongs to family A in the genealogical report of Drugge et al.3 All patients had a history of lifelong exercise intolerance with symptoms of muscle fatigue, shortness of breath, and tachycardia with minor levels of physical exertion, associated with episodes of myoglobinuria. Physiological investigations revealed low work (0.3–0.5 W/kg, normal mean 2–3 W/kg) and oxidative capacity (10 to 12 ml/kg/min, normal mean ± standard deviation [SD], 39 ± 5 ml/kg/min) in all patients, together with a marked deficiency of succinate dehydrogenase (SDH, 0.5 to 0.8 μmol/min per g tissue, normal mean ± SD 1.8 ± 0.5 μmol/min per g tissue) and aconitase (1.9 to 3.1 μmol/min per g tissue, normal mean ± SD 7.8 ± 2.0 μmol/min per g tissue) in the patients' muscle. The unaffected offspring, H1, had normal results of both physiological and biochemical investigations. We obtained blood samples and skin and muscle biopsies from the three affected individuals and the unaffected offspring after they had given written consent in accordance with the Institutional Review Board for Human Studies of the University of Texas, Southwestern Medical School, which approved the experimental protocol. Genomic DNA was extracted from leukocytes via standard procedures. Skin biopsy was performed on the volar forearm with a 4 mm diameter skin-biopsy punch (Miltex), and fibroblasts were cultured in fetal calf serum. Vastus lateralis muscle biopsies were performed with a 6G Bergstrom needle (Popper). Muscle samples were snap frozen and stored in liquid nitrogen until analyzed biochemically, or they were frozen in 2-methylbutane (isopentane), precooled to approximately −150°C in liquid nitrogen, and then stored at −80°C or in liquid nitrogen for histochemistry.
SNP Microarray Genotyping and Mutation Detection
The disease was assumed to be recessive with homozygosity by descent. The four individuals were genotyped with Infinium HumanHap550 SNP genotyping chips, which contain 555,352 unique SNPs, as per the manufacturer's instructions (Illumina). An Illumina BeadStation scanner and data-collection software were used to collect the data. BeadStudio's genotyping module (v2.3.25, Illumina) was used to generate genotypes. Genome viewer, a visualization tool from BeadStudio, was then used to investigate the log R ratio and the B allele frequencies of the SNPs.
The candidate gene ISCU was PCR amplified and sequenced in the four genomic DNA samples by dye-terminator sequencing (BigDye version 3.1; Applied Biosystems). For the ISCU genomic DNA amplification, primer sets were designed to cover the entire sequence and 2 kb upstream of the ISCU gene (primer sequences and conditions used in the sequencing analysis are available upon request). Sequences were aligned and compared with consensus database sequences with Sequencher 4.7 software (Gene Codes Corporation).
RNA Analysis
RNA was extracted from muscle of the three affected and unaffected individuals and two unrelated controls. Total RNA was isolated from 50 mg of muscle tissue with the QIAGEN RNeasy Fibrous Tissue Kit as per the manufacturer's instructions. Fibroblasts from two affected homozygotes, the heterozygote, and a control were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 20% fetal bovine serum, 2 mM L-glutamine, and antibiotics. The cells were then harvested for RNA and protein isolation and quantification. TRIzol reagent (Invitrogen) was used to isolate total RNA from the fibroblasts, and the RNeasy Mini kit (QIAGEN) was used to purify the RNA. The total RNA from muscle and fibroblasts was then converted to cDNA by the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Specific primers and probes (Table 1) were designed with the Primer Express Program (Applied Biosystems) and used to amplify the ISCU mRNA. Primers to amplify the internal control human beta-glucuronidase (HGUSB) were purchased from Applied Biosystems. Quantitative reverse transcriptase PCR (RT-PCR) reactions were run in triplicate, and the experiments were performed in triplicate with the ABI Prism 7900 Sequence Detector System (Applied Biosystems). The PCR cycling conditions were standard 95°C for 10 min for one cycle, 95°C for 15 s, and 60°C for 1 min for 40 cycles. The level of each transcript was quantified by the cycle at which the PCR amplification was in log phase where there was significant fluorescent signal (Ct) with HGUSB as the endogenous control. The values were normalized to the mean of the unrelated controls via the 2-ΔΔCt method. RT-PCR reactions were used to analyze possible splice variants. The following ISCU-specific primers were used: 5′-GATATCGCCAAGGAGCTCTG-3′ (RT-PCR 5-5.2F, Exon 4) and the antisense primers 5′-GACCAAACTGGAAAGCCAAG-3′ (RT-PCR 5-5.2R, Exon 4A) and 5′-TCATTTCTTCTCTGCCTCTCC-3′ (WHT 2′ R, Exon 5). Each RT-PCR reaction was done in a total reaction volume of 15 μl containing the following: 12 μl of FastStart PCR Master Mix (Roche Applied Science), 1 μl of each primer (10 pmol/μl), and 1 μl of cDNA solution. The cycling parameters were as follows: 95°C for 10 min to denature the DNA; 10 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s; 20 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s; and then one cycle of 72°C for 10 min. The RT-PCR reactions were analyzed on a 2% agarose gel to characterize alternate splicing.
Table 1.
Quantitative RT-PCR Primer and Probe Sequences Used to Amplify and Quantitate the ISCU Transcripts
| ISCU Exon | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Probe |
|---|---|---|---|
| 1A/2 | CCCCGGCCCGACTCTAT | CAGTCCAGTTCCAACATTTTTAGATG | AAGGTTGTTGATCATTATGAAAATCCTAGAAACG |
| 1A/1B | CCCCGGCCCGACTCTA | TTTTGTGTCTCCTCTACAATACTTCACA | CACAAGAAGGTATCTCAAA |
| 2/3 | GTGGTGACGTAATGAAATTACAGATTC | TAATGAGCTGGAGGCAATTGC | TGGATGCTAGGTTTAAAACATTTGGCTGTGGTT |
| 3/4 | CCACTGAATGGGTGAAAGGAA | GCAGAGCTCCTTGGCGATAT | CGGTGGAGGAAGCCTTGACTATCAAAAACA |
| 4/5 | TGCCTTCCTCCCGTGAAAC | CCAGGGCGGCCTTGA | ACTGCTCCATGCTGGCTGAAGATGC |
Aconitase Activity and Western Blotting
Muscle lysates were prepared in an anaerobic chamber to minimize loss of enzyme activity and degradation of oxygen-sensitive proteins. Frozen muscle samples were homogenized with a mortar and pestle chilled with liquid nitrogen and were lysed in argon-saturated lysis buffer containing 25 mM Tris-Cl (pH 7.5), 40 mM KCl, 1% Triton, 2 mM citrate, 1 mM DTT, 0.6 mM MnCl2, 1 mM AEBSF, and complete TM EDTA-free protease inhibitor cocktail (Roche Applied Science). The mitochondrial and cytosolic aconitases were separated from total cellular extracts electrophoretically on nondenaturing PAGE, and the aconitase activity was performed as previously described.7 The antibodies used in these studies were as follows: rabbit polyclonal anti-IRP1,7 rabbit polyclonal anti-m-aconitase,7 and monoclonal anti-tubulin (Sigma). For ISCU, we used rabbit polyclonal antibody,8 for which the epitope is in the middle of the protein (KLQIQVDEKGKIVDARFK) and mostly encoded by exon 3.
Histochemistry Studies
Unfixed frozen muscle sections were serially cut at 10 μm thickness, applied to cover glasses or glass-plus slides, and air-dried. SDH stains were performed immediately after sectioning as previously described.9 Serial sections reserved for iron staining were wrapped in foil and stored at −80°C. Sections were thawed just prior to staining. Perl's Prussian Blue stain for ferric iron was adapted from a protocol previously described.7 Muscle sections were fixed in 10% neutral buffered formalin solution for 10 min, followed by rinsing in Millipore Milli-Q Plus water for 5 min. Sections were then incubated at room temperature in Perl's solution (freshly made aqueous solution of 2% potassium ferrocyanide and 2% HCl). Sections were then rinsed in Millipore Milli-Q Plus running water for 5 min, followed by incubation in DAB Stain (1.0 M Tris-HCl buffer, pH 7.5 [40 ml], with 3% H202 [1 ml] and 30 mg DAB) for 30 min at room temperature. Slides were rinsed in running tap water for 5 min. They were then dehydrated in alcohol (95% × 2; 100% × 3), cleared in xylenes (× 3), and mounted with Cytoseal mounting medium. Photomicrographic images were observed with a Nikon FXA Microphot and were captured with a Nikon DXM 1200F camera.
Results
We used single-nucleotide polymorphism (SNP) microarray genotyping for homozygosity mapping in three affected patients and one patient's unaffected offspring in three separate families from northern Sweden. The SNP analysis showed shared homozygosity in the affected individuals in a 2.7 Mb segment of chromosome 12q (Figure 1). Haplotypes were conserved across the interval of homozygosity, consistent with a common origin (Figure 1). This interval contains 43 known genes and two open reading frames, among which the iron-sulfur-cluster assembly enzyme ISCU was the most likely candidate in light of the biochemical defect in the patient muscle. ISCU is believed to be the primary scaffold upon which iron-sulfur clusters are assembled, and it has two known isoforms in humans, cytosolic and mitochondrial, resulting from alternative splicing near the 5′ end of the pre-mRNA.8,10 These isoforms are thought to coordinately drive the reprogramming of iron metabolism in different subcellular compartments to regulate overall cellular iron trafficking in response to iron needs.7 We sequenced all exons and introns, the 3′UTR, and 2 kb upstream of the ISCU gene to cover the likely promoter region. We found a single intronic change at position 107,485,556 bp of chromosome 12, hereafter named g.7044 G→C (GenBank accession number EU334585), homozygous in all patients and heterozygous in the unaffected offspring (Figure 2). We analyzed 568 chromosomes from controls originating from the same region in northern Sweden as the patients and found only three heterozygous changes of this nucleotide, resulting in an allele frequency of approximately 1/188 in this population. We also analyzed 584 chromosomes from non-Swedish European and American controls and found no change of this nucleotide.
Figure 1.
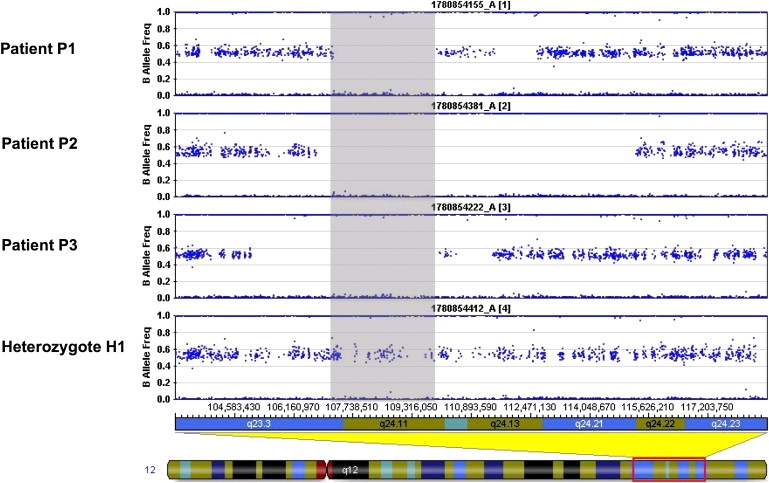
Homozygosity Mapping by SNP Microarray in the Swedish Families
Homozygosity mapping by SNP array in the three northern Swedish families with myopathy and deficiency of succinate dehydrogenase and aconitase. Each panel shows results from the affected individuals (patients P1 to P3) and the unaffected family member (individual H1). The horizontal band in each panel represents heterozygous signal from two-allele SNP markers distributed along chromosome 12. The gray shaded box indicates a region of homozygosity (loss of heterozygous signal) at 12q shared by all three affected individuals and not the unaffected son H1 of affected individual P3.
Figure 2.
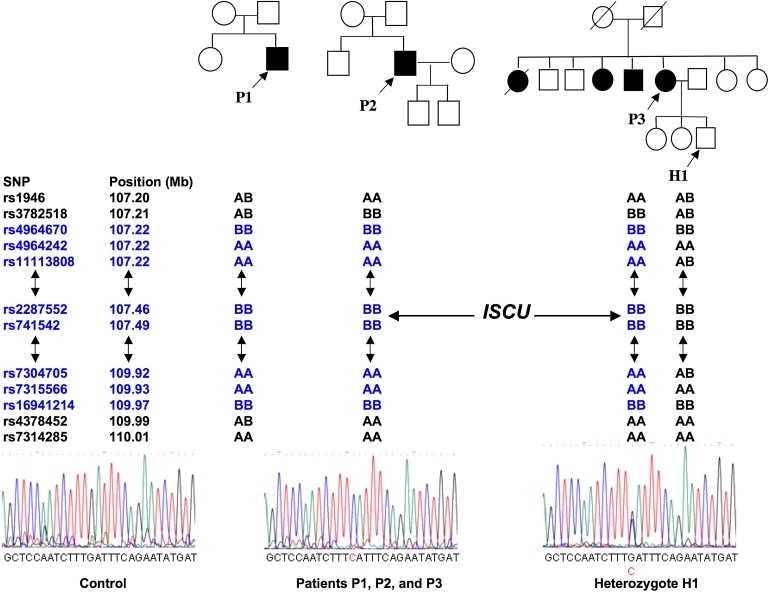
SNP-Array Genotyping and Mutation Detection in the Swedish Families
Pedigrees and electropherograms of the ISCU mutation (g.7044 G→C) in the three families from northern Sweden, and SNP alleles flanking the ISCU gene. The positions of the SNP markers on chromosome 12 are indicated in Mb, with the corresponding alleles designated by A and B. AA or BB indicates that the proband is homozygous for SNP markers that surround the ISCU gene (the position is shown by an arrow). Double-headed arrows indicate a long stretch of homozygous markers. All patients share the same homozygous haplotype (highlighted in blue), consistent with a common origin.
Because this mutation replaces a repressive G with a C and extends an intronic polypyrimidine tract in intron 4 (to TCTTTC instead of TCTTTG),11 we hypothesized that it induced abnormal splicing of the ISCU pre-mRNAs. The splice prediction tool of Berkeley Drosophila Genome Project confirmed that the genomic position at which we found the homozygous change is located within the polypyrimidine tract of a potential splice acceptor site, which would promote spliceosome assembly. The probability score for this splice acceptor site to be functional would be strengthened from 0.87 (on a scale from 0 to 1) to 0.95, as a result of the single-nucleotide substitution found in the Swedish patients. Similar results were obtained when other splice prediction tools were used, e.g., NetGene2 server. RT-PCR performed in patient and control muscle with subsequent cDNA sequencing revealed the existence of a previously unrecognized ISCU transcript containing an additional exon (exon 4A, GenBank accession number EU329002), upstream of the known terminal exon (Figures 3 and 4A). Quantitative RT-PCR performed in patient muscle confirmed that the retention of exon 4A in the ISCU mRNA is markedly enhanced compared to controls (data not shown). With the homozygous mutation, the ISCU transcripts containing exon 4A are more abundant in patient muscle compared to controls (Figure 4A). In addition, qRT-PCR revealed a decrease in the total ISCU mRNA levels in patient muscle; exon 1A-2, 1A-1B, 2-3, and 3-4 levels were 38%–46%, 18%–44%, 40%–46%, and 35%–43% of controls, respectively, whereas exon 4-5 levels were 7%–8% of controls. It is possible that the stability of the ISCU mRNA is altered in skeletal muscle because of the inclusion of the Swedish myopathy exon, which would introduce a premature stop codon 53 bp upstream from the last exon junction and therefore possibly activate nonsense-mediated decay (NMD) (Figure 3). However, we could not test this hypothesis because the level of ISCU mRNA in cultured fibroblasts was normal. Accordingly, when we added cycloheximide, mRNA levels from patient fibroblasts did not change. Use of cycloheximide in cultured muscle cells from patients could potentially allow insight into why mRNA levels decrease in the skeletal muscle of these patients, where NMD may be selectively activated.12 Further studies in patient muscle indicate the existence of additional transcripts also containing sequences of the exon 4A (Figure 4A). However, these transcripts are in very low abundance, and their rare presence in control muscle as well (data not shown) strongly argues against their having a role in the disease.
Figure 3.
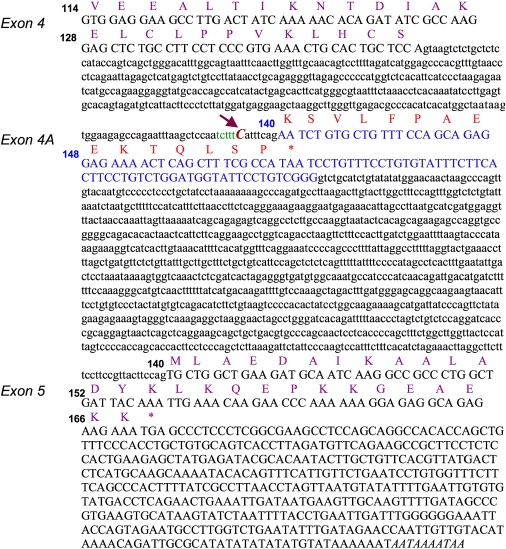
Sequences of the Splice Mutation and the Additional Exon 4A
The intronic Swedish myopathy mutation, and exon 4A. Genomic sequences of exons 4 and 5 with the corresponding protein residues indicated in purple. The intronic mutation in patients (g.7044 G→C), indicated by an arrow, extends a polypyrimidine tract (in green) and leads to the inclusion of an additional exon 4A (in blue) that is predicted to result in a premature stop codon (marked as ∗) 53 bp upstream from the last exon junction. Numbers indicate ISCU protein-codon residues.
Figure 4.
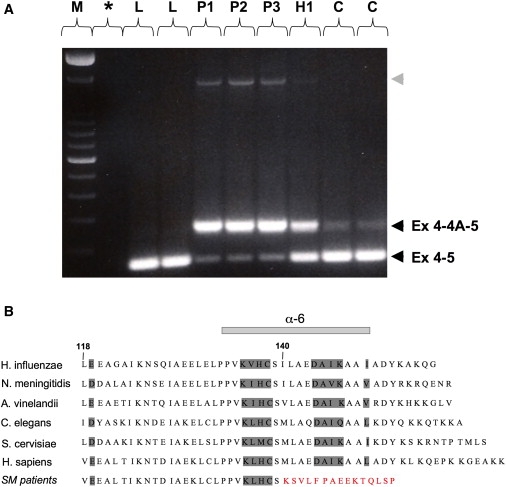
Abnormal ISCU Transcripts and Predicted Alteration of the Peptide Sequence
(A) RT-PCR between exons 4 and 5, showing two different transcripts in patients and controls, the normal transcript containing exons 4 and 5, and one containing exons 4, 4A, and 5, as confirmed by cDNA sequencing. The retention of exon 4A is markedly enhanced in patient muscle. Both transcripts are present in the heterozygote. Note the presence of an additional larger transcript in patient muscle (gray arrowhead), for which cDNA sequencing revealed the presence of exons 4, 4A, and 5, as well as the retention of the intronic sequence between exon 4A and exon 5. These transcripts are in very low abundance, and their rare presence in control muscle as well (data not shown) strongly argues against their having a role in the disease. L indicates cDNA from lymphoblastoid cells from controls; P, H, and C indicate cDNA from muscle of the three northern Swedish patients, the heterozygote, and controls, respectively; M indicates 100 bp DNA ladder (Invitrogen); and ∗ indicates no cDNA control.
(B) Alignment of residues encoded by exons 4 and 5, showing the strong conservation of ISCU among species, as well as the alteration of the C terminus of ISCU resulting from the splice mutation in patients (SM). Numbers designate ISCU protein-codon residues, and the residues corresponding to the α6-helix are indicated.
Consistent with a deleterious effect of the mutation on the ISCU protein, western-blot analysis of patient muscle showed a substantial reduction of the normal mitochondrial isoform (m-ISCU) (Figure 4A), in agreement with the major reduction of normal transcript containing exons 4 and 5 in patient muscle (to 7%–8% compared of controls). Furthermore, the mutation is predicted to alter the C terminus of ISCU, especially the highly conserved residues that are within the sixth α-helix (Figure 4B). Because this structural alteration is in the proximity of one of the invariant cysteine residues that are involved in the coordination of the iron-sulfur cluster,13 it would probably diminish the ability of the mutant protein to assemble and deliver iron-sulfur cluster to target proteins. Given that the epitope for the ISCU antibody recognizes a part of the protein that is mostly encoded by exon 3, both wild-type and mutant proteins would be recognized. However, the western blot did not detect any mutant m-ISCU. This may be explained by the following: (1) the activation of NMD in patient muscle as a result of the premature stop codon and consistent with the overall reduction of ISCU transcripts (40% of controls on average); (2) the splicing defect resulting in an alteration of the C terminus of ISCU, a change that could affect the stability of mutant peptide or its ability to dimerize properly;14 and (3) a dominant-negative effect of the mutant peptide on the stability and functioning of the normal peptide.
We analyzed the functional consequences of low muscle m-ISCU on iron-sulfur proteins and intracellular iron homeostasis in patient muscle. Activities of both mitochondrial aconitase and the bifunctional cytosolic aconitase/iron regulatory protein 1 (IRP1)15 were decreased in the patient muscle (Figure 5B). Supporting previous biochemical findings, the protein levels of mitochondrial aconitase were reduced in patients compared to controls (Figure 5A). The protein levels of the iron-sensing protein IRP1 were also low in patient muscle (Figure 5B), suggesting a destabilization of these iron-sulfur proteins as a result of the loss of iron-sulfur clusters.16 Addition of a reductant such as β-mercaptoethanol activates the binding of IRP1 to transcripts containing iron-responsive elements (IREs) by converting cytosolic aconitase to the IRE-binding form. Gel-shift assays showed that the amount of activatable IRP1 was reduced in patients compared to controls, consistent with the decrease in aconitase activity (Figure 5C). In addition, histochemical studies showed a notable increase in iron staining in patient muscle fibers compared to controls (Figure 6). This iron overload was observed exclusively in SDH-negative fibers, where m-ISCU is likely to be most depleted (Figures 6A and 6B). However, we found no evidence of fiber-type specificity with respect to SDH-negative, iron-overloaded fibers. The punctate distribution of the iron staining in patients with this myopathy with deficiency of succinate dehydrogenase and aconitase is consistent with the mitochondrial iron overload previously detected in ultrastructural studies of these patients.5 This punctate ferric-iron staining is also reminiscent of the iron deposition pattern observed in cells depleted of m-ISCU by siRNA silencing,7 emphasizing the downstream effect of the splice mutation on mitochondrial iron homeostasis in the muscle of these patients. Interestingly, all biochemical and histological studies were normal in the heterozygous individual (Figures 5 and 6), consistent with a gene-dosage effect in this disease. To confirm that deficiency of m-ISCU is not a nonspecific effect of mitochondrial respiratory-chain defects, we analyzed muscle from a patient with a mitochondrial myopathy due to a single large mitochondrial DNA deletion, which was abundant in the skeletal muscle. This patient's muscle had normal aconitase activity (Figure 5B) and normal SDH and iron staining (data not shown), as well as a relative increase (rather than a decrease) in m-ISCU levels (Figure 5A).
Figure 5.
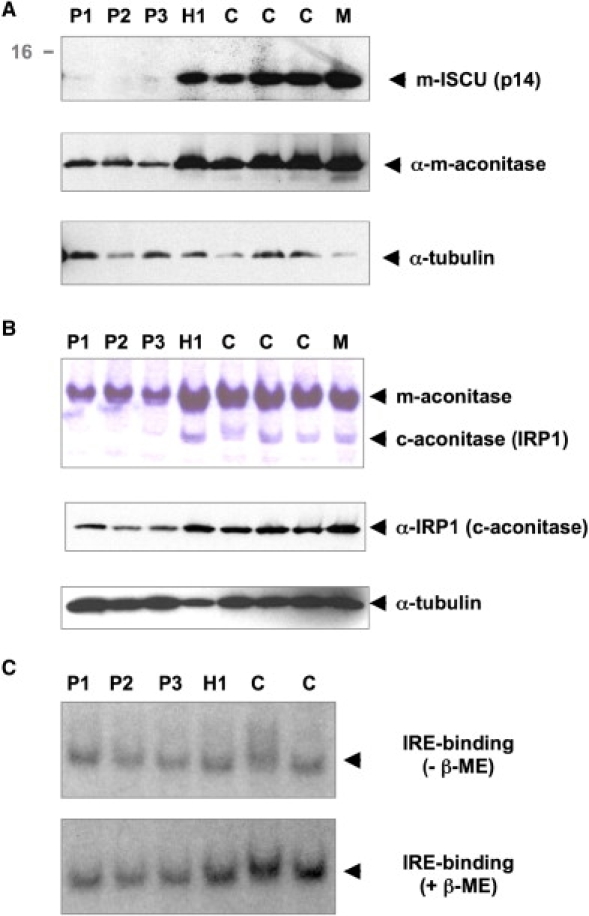
Mitochondrial ISCU and Iron-Sulfur-Cluster-Containing Proteins in Skeletal Muscle
Mitochondrial ISCU (m-ISCU) and iron-sulfur-cluster-containing proteins in muscle from northern Swedish patients (P1–P3), the unaffected offspring (H1), controls (C), and a patient with a myopathy due to a mitochondrial DNA deletion (M).
(A) Western blots of m-ISCU and mitochondrial aconitase show a marked reduction in patient muscle. Note the increased m-ISCU level in the control patient with mitochondrial myopathy.
(B) Aconitase enzymatic activity was determined after separation of cellular extracts in nondenaturing PAGE. The fast band corresponds to cytosolic aconitase (c-aconitase), and the slower band corresponds to the mitochondrial isoform (m-aconitase). The activities of both c- and m-aconitase are deficient in patients. Western blots of the iron-regulatory protein IRP1 also show reduced protein levels in patient muscle, consistent with the decrease in c-aconitase activity.
(C) Activation of IRP1 to iron-responsive elements (IREs) with β-mercaptoethanol in skeletal muscles from northern Swedish patients (P1–P3), the unaffected offspring (H1), and controls (C). β-mercaptoethanol activates the binding of IRP1 to transcripts containing IRE by converting cytosolic aconitase to the IRE-binding form. There is no activation of IRE-binding in patients in contrast to controls, consistent with the decrease in aconitase activity in the patient samples.
Figure 6.
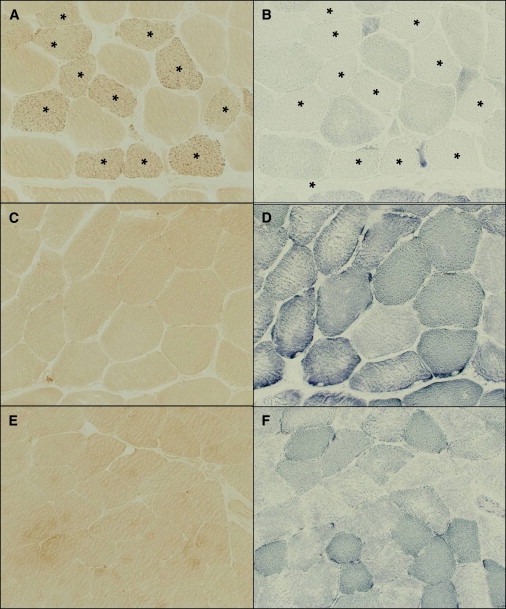
SDH and Iron Staining in Skeletal Muscle
Histochemistry of skeletal muscle from a northern Swedish patient (A and B), the heterozygote (C and D), and a control (E and F). Serial sections were stained for ferric iron (A, C, and E) and SDH (B, D, and F). Note the presence of intracellular iron overload in SDH-deficient fibers (marked with ∗) in the patient sample. The punctate distribution of the iron staining is consistent with the mitochondrial iron overload previously detected in ultrastructural studies of these patients.5
Discussion
We have shown that myopathy with deficiency of succinate dehydrogenase and aconitase is an autosomal-recessive metabolic disease caused by mutations in the ISCU gene. The splice mutation detected in patients from northern Sweden results in aberrant splicing, with the increased retention of an additional exon and the introduction of a premature stop codon in the penultimate exon; this ultimately alters the C terminus of the protein and decreases levels of ISCU mRNA and protein. The depletion of mitochondrial ISCU in muscle accounts for the biochemical and clinical phenotype, which is characterized by a deficiency in mitochondrial iron-sulfur proteins and impaired muscle oxidative metabolism.
Iron-sulfur clusters are prosthetic groups composed of iron and sulfur and are usually ligated to proteins via the sulfhydryl side chains of cysteine. Iron-sulfur clusters often function as electron acceptors or donors, and they are important for function of the mitochondrial respiratory chain. In humans, there are twelve known iron-sulfur clusters in respiratory complexes I–III. In addition to their importance in electron transfer, iron-sulfur clusters can ligate substrate in enzymes such as aconitase, which converts citrate to isocitrate, and iron-sulfur proteins can also have important structural and sensing roles.10 In mammalian iron-sulfur-cluster assembly, a cysteine desulfurase known as ISCS provides sulfur, and assembly of nascent iron-sulfur clusters takes place on ISCU, which functions as a scaffold upon which the cluster is assembled.17 Frataxin is thought to participate in iron-sulfur-cluster assembly by binding ferrous iron to acidic residues18 and thereby maintaining iron in a nonaggregated bioavailable form. In addition, frataxin physically interacts with ISCU, and this interaction probably facilitates delivery of iron from frataxin to nascent iron-sulfur clusters on ISCU.19
The loss of the mitochondrial protein frataxin in Friedreich's ataxia is associated with reduced activity of various mitochondrial iron-sulfur proteins, including aconitase,20,21 and loss of frataxin eventually causes mitochondrial iron accumulation in the central nervous system and heart.10,20 Also, loss of glutaredoxin 5, which is important in iron-sulfur-cluster assembly,22 results in sideroblastic anemia in humans with a glutaredoxin 5 splicing defect.23 Of note, the defining characteristic of sideroblastic anemias is mitochondrial iron overload. Similarly, ISCU depletion results in abnormal iron staining, both in the muscle of patients with this myopathy5 (Figure 6) and in cell culture when ISCU levels are reduced by RNAi techniques.7 Swedish myopathy therefore represents another example in which decreased ability to synthesize iron-sulfur clusters leads to mitochondrial iron overload. This observation supports the possibility that one or more iron-sulfur-cluster proteins act as sensors for regulation of mitochondrial iron homeostasis.10 ISCU is now the third iron-sulfur-cluster assembly protein implicated in a human disease, along with frataxin and glutaredoxin 5. The interaction of frataxin with ISCU and the biochemical and histological features associated with this ISCU mutation also indicate that the myopathy is caused by defects in iron-sulfur cluster assembly and intracellular iron metabolism similar to Friedreich ataxia, but with different tissue specificity. It has been recently suggested that the downregulation of ISCU in frataxin-deficient cells further aggravates the deficiency of iron-sulfur proteins in Friedreich ataxia.24,25 The novel implication of ISCU in a human disorder may therefore contribute to a better understanding of the role of frataxin in mammalian iron-sulfur cluster assembly.
In addition, this myopathy with succinate dehydrogenase and aconitase deficiency is an interesting example of a mutation that strengthens a weak splice acceptor site, resulting in exon retention and the subsequent overexpression of a deleterious isoform. Only a few examples of this mutational mechanism have been reported previously.26,27 Alternative splice variants with aberrant exon inclusion may also cause tissue-specific pathology due to the tissue-specific expression of partner proteins.28 The identification of the disease-causing mutation in this metabolic muscle disease may offer new therapeutic options for the affected patients, for example through modulation of intracellular iron metabolism29 or correction of the aberrant splicing with antisense oligonucleotides.30
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html
NetGene2 server, http://www.cbs.dtu.dk/services/NetGene2/
Accession Numbers
The splice mutation and the additional exon 4A sequences reported in this paper have been deposited to the GenBank under the EU334585 and EU329002 accession numbers, respectively.
Acknowledgments
The authors wish to thank Nadine Romain, George Harmison, and Ian Rafferty for their help with the preparation of muscle tissues, the fibroblast culture, and the SNP-microarray genotyping, respectively, and Marguerite Gunder for invaluable assistance. We thank Drs. Karl G. Henriksson and Bjorn Lindvall for identifying and referring the patients. This work was supported by the intramural program of the National Institute of Neurological Disorders and Stroke, the National Institute on Aging, and the National Institute of Child Health and Human Development and by grants from the Muscular Dystrophy Association, the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS) (R01 AR050597), and a Veterans Affairs (VA) Merit Review (R.G.H.). M.A.K. was supported by a National Institute of Neurological Disorders and Stroke (NINDS) Competitive Postdoctoral Fellowship.
References
- 1.Larsson L.E., Linderholm H., Mueller R., Ringqvist T., Soernaes R. Hereditary metabolic myopathy with paroxysmal myoglobinuria due to abnormal glycolysis. J. Neurol. Neurosurg. Psychiatry. 1964;27:361–380. doi: 10.1136/jnnp.27.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linderholm H., Muller R., Ringqvist T., Sornas R. Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Med. Scand. 1969;185:153–166. doi: 10.1111/j.0954-6820.1969.tb07314.x. [DOI] [PubMed] [Google Scholar]
- 3.Drugge U., Holmberg M., Holmgren G., Almay B.G., Linderholm H. Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: A genealogical study. J. Med. Genet. 1995;32:344–347. doi: 10.1136/jmg.32.5.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linderholm H., Essen-Gustavsson B., Thornell L.E. Low succinate dehydrogenase (SDH) activity in a patient with a hereditary myopathy with paroxysmal myoglobinuria. J. Intern. Med. 1990;228:43–52. doi: 10.1111/j.1365-2796.1990.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 5.Haller R.G., Henriksson K.G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N.H., Gunder M., Ayyad K., Blomqvist C.G. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 1991;88:1197–1206. doi: 10.1172/JCI115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall R.E., Henriksson K.G., Lewis S.F., Haller R.G., Kennaway N.G. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 1993;92:2660–2666. doi: 10.1172/JCI116882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong W.H., Rouault T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Tong W.H., Rouault T. Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 2000;19:5692–5700. doi: 10.1093/emboj/19.21.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen H., Hoyer P.E. Studies in succinate dehydrogenase histochemistry. Histochemie. 1973;35:173–188. doi: 10.1007/BF00306043. [DOI] [PubMed] [Google Scholar]
- 10.Rouault T.A., Tong W.H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- 11.Singh R., Valcarcel J., Green M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 12.Bateman J.F., Freddi S., Nattrass G., Savarirayan R. Tissue-specific RNA surveillance? Nonsense-mediated mRNA decay causes collagen X haploinsufficiency in Schmid metaphyseal chondrodysplasia cartilage. Hum. Mol. Genet. 2003;12:217–225. doi: 10.1093/hmg/ddg054. [DOI] [PubMed] [Google Scholar]
- 13.Liu J., Oganesyan N., Shin D.H., Jancarik J., Yokota H., Kim R., Kim S.H. Structural characterization of an iron-sulfur cluster assembly protein IscU in a zinc-bound form. Proteins. 2005;59:875–881. doi: 10.1002/prot.20421. [DOI] [PubMed] [Google Scholar]
- 14.Agar J.N., Krebs C., Frazzon J., Huynh B.H., Dean D.R., Johnson M.K. IscU as a scaffold for iron-sulfur cluster biosynthesis: Sequential assembly of [2Fe-2S] and [4Fe-4S] clusters in IscU. Biochemistry. 2000;39:7856–7862. doi: 10.1021/bi000931n. [DOI] [PubMed] [Google Scholar]
- 15.Rouault T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Fillebeen C., Chen G., Biederbick A., Lill R., Pantopoulos K. Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2007;27:2423–2430. doi: 10.1128/MCB.01111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandramouli K., Unciuleac M.C., Naik S., Dean D.R., Huynh B.H., Johnson M.K. Formation and properties of [4Fe-4S] clusters on the IscU scaffold protein. Biochemistry. 2007;46:6804–6811. doi: 10.1021/bi6026659. [DOI] [PubMed] [Google Scholar]
- 18.Bencze K.Z., Kondapalli K.C., Cook J.D., McMahon S., Millan-Pacheco C., Pastor N., Stemmler T.L. The structure and function of frataxin. Crit. Rev. Biochem. Mol. Biol. 2006;41:269–291. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan Y., Napoli E., Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- 20.Rotig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 21.Bulteau A.L., O'Neill H.A., Kennedy M.C., Ikeda-Saito M., Isaya G., Szweda L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 22.Wingert R.A., Galloway J.L., Barut B., Foott H., Fraenkel P., Axe J.L., Weber G.J., Dooley K., Davidson A.J., Schmid B. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 23.Camaschella C., Campanella A., De Falco L., Boschetto L., Merlini R., Silvestri L., Levi S., Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 24.Tan G., Napoli E., Taroni F., Cortopassi G. Decreased expression of genes involved in sulfur amino acid metabolism in frataxin-deficient cells. Hum. Mol. Genet. 2003;12:1699–1711. doi: 10.1093/hmg/ddg187. [DOI] [PubMed] [Google Scholar]
- 25.Martelli A., Wattenhofer-Donze M., Schmucker S., Bouvet S., Reutenauer L., Puccio H. Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum. Mol. Genet. 2007;16:2651–2658. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- 26.Malkani R., D'Souza I., Gwinn-Hardy K., Schellenberg G.D., Hardy J., Momeni P. A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol. Dis. 2006;22:401–403. doi: 10.1016/j.nbd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi P., Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rump A., Rosen-Wolff A., Gahr M., Seidenberg J., Roos C., Walter L., Gunther V., Roesler J. A splice-supporting intronic mutation in the last bp position of a cryptic exon within intron 6 of the CYBB gene induces its incorporation into the mRNA causing chronic granulomatous disease (CGD) Gene. 2006;371:174–181. doi: 10.1016/j.gene.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 29.Boddaert N., Le Quan Sang K.H., Rotig A., Leroy-Willig A., Gallet S., Brunelle F., Sidi D., Thalabard J.C., Munnich A., Cabantchik Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood. 2007;110:401–408. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Blanco M.A., Baraniak A.P., Lasda E.L. Alternative splicing in disease and therapy. Nat. Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]