

Abstract
Prediction of protein-RNA interactions at the atomic level of detail is crucial for our ability to understand and interfere with processes such as gene expression and regulation. Here, we investigate protein binding pockets that accommodate extruded nucleotides not involved in RNA base pairing. We observed that most of the protein interacting nucleotides are part of a consecutive fragment of at least two nucleotides, whose rings have significant interactions with the protein. Many of these share the same protein binding cavity and more than 30% of such pairs are π-stacked. Since these local geometries can not be inferred from the nucleotide identities, we present a novel framework for their prediction from the properties of protein binding sites. First, we present a classification of known RNA nucleotide and dinucleotide protein binding sites and identify the common types of shared 3D physico-chemical binding patterns. These are recognized by a new classification methodology which is based on spatial multiple alignment. The shared patterns reveal novel similarities between dinucleotide binding sites of proteins with different overall sequences, folds and functions. Given a protein structure, we use these patterns for the prediction of its RNA dinucleotides binding sites. Based on the binding modes of these nucleotides, we further predict an RNA fragment that interacts with those protein binding sites. With these knowledge-based predictions we construct an RNA fragment that can have a previously unknown sequence and structure. In addition, we provide a drug design application in which the database of all known small molecule binding sites is searched for regions similar to nucleotide and dinucleotide binding patterns, suggesting new fragments and scaffolds that can target them.
Keywords: protein-RNA interactions, nucleotide and dinucleotide binding sites, physico-chemical binding patterns, multiple binding site alignment, RNA aptamer drug design
1 Introduction
Protein-RNA interactions are crucial for many cellular processes, such as gene expression and regulation as well as protein synthesis. Understanding and predicting such interactions at the atomic level is crucial for our ability to interfere with malfunctioning processes in disease. Consequently, analysis of the protein-RNA interactions and RNA binding sites have been a field of intensive research. The pioneering works of Jones et al.1 and Nadassy et al2 have provided important insights into the physico-chemical and geometrical nature as well as the amino acid composition of these regions. The specific atomic interactions formed between nucleotides and amino acids have been analyzed and compared in several important contributions 3–8. By analyzing the amino acid composition in RNA binding sites, several successful methods for prediction of RNA binding sites were developed 9–13.
However, most of these prediction methods do not distinguish between two main interaction types which were thoroughly studied by Draper14: (1) interactions with the backbone of double-stranded RNA molecules; (2) interactions of single-stranded RNA bases that are accommodated in the protein binding pockets. Due to the differences between the two types their prediction should be addressed separately. As noted in a recent comprehensive review of Auweter et al15, while the first type of interactions occur through positively charged protein surface patches, the second type of contacts with single-stranded nucleotides, often involve hydrophobic patches. The contacts are often with the unpaired nucleic acid bases, while the direct contacts with the phosphate moieties of the backbone, which point towards the bulk solution can be rare15. Here, we focus on the prediction of protein interactions formed with the single-stranded nucleotide bases. Sequences of such nucleotides, which are not involved in local base pairs and are extruded from the surrounding double-stranded helix, also termed extruded helical single strands, were recently described as special motifs in SCOR (Structural Classification of RNA) and were proposed to be mediators in RNA-RNA and RNA-protein interactions16, 17.
Several works have classified the protein-RNA interactions based on the sequences and/or overall structures of the corresponding protein18–20 or RNA molecules17, 21, 22. Sykes and Levitt have classified all doublets of spatially close nucleotides23. However, these do not always capture the similarity in the local regions which are responsible for protein-RNA binding. Analysis of these regions is important due to several reasons. First, proteins of the same family can form different interactions with RNA nucleotides 24–26. Second, RNA molecules can be flexible and can explore different conformations that are “fixed” by the protein whose binding site is more rigid and quenches this motion27, 28. This observation is also supported by a recent study of Ellis and Jones29 who evaluated the conformational changes in known RNA binding proteins and observed that the flexibility in the protein binding sites is not significant and should allow the structural prediction of these interaction regions. Previous works that analyzed the nucleotides’ physico-chemical binding patterns focused on single nucleotides1, 5, 30, 31. Moreover, to the best of our knowledge, the results of their studies have never been applied to atomic level prediction of protein-RNA interactions and RNA structures.
Here, we investigate the protein binding pockets that accommodate extruded nucleotides not involved in RNA base pairing. We observed that many of these protein cavities are common to pairs of consecutive nucleotides that are often π-stacked with each other. Consequently, we suggest that consideration of binding patterns of pairs of consecutive RNA nucleotides may be essential for the correct prediction of protein-RNA interactions. We observed that the local nucleotide geometries can not be inferred from the nucleotide identities which led us to develop a novel framework for their prediction through the recognition of known protein binding patterns.
Specifically, we present a classification of nucleotide and dinucleotide binding sites, which are described by a set of physico-chemical properties that may be created by amino acids with different identities and spatial location of backbone atoms. Toward this goal, we have developed a new classification algorithm which performs multiple structural alignments and validates the spatial superimposition of the cluster members. The created clusters describe the common types of 3D consensus binding patterns that are used for several applications. First, by searching for these patterns on the surface of a complete protein, we predict its potential dinucleotide binding sites at the atomic level of detail. Second, using the binding modes of the nucleotides bound to the 3D patterns, we further predict a protein interacting RNA fragment. Finally, we suggest that searching the database of drug binding sites for patterns similar to nucleotide and dinucleotide binding sites can assist in the prediction of ligands and ligand fragments that can be used to interfere with protein-RNA interactions.
2 Results
Our goal is to recognize and predict the main types of interactions between protein binding sites and single-stranded RNA bases. Specifically, we focus on the protein binding pockets that accommodate extruded16 nucleotides not involved in RNA base pairing (see Figure 1). We define a nucleotide binding site by the protein Connolly solvent accessible surface area32 within 2Å from the surface of the RNA nucleotide ring. Nucleotides with a protein binding site area larger than 3Å2 are considered as protein interacting. Given a pair of extruded consecutive nucleotides that interact with the protein, a dinucleotide binding site is defined by the pair of the corresponding nucleotide binding sites. The physico-chemical properties of the binding sites are represented by points in 3D space termed pseudocenters, extracted from the protein amino acids according to Schmitt et al33. Each pseudocenter represents a group of atoms according to the interactions in which it may participate: hydrogen-bond donor, hydrogen-bond acceptor, mixed donor/acceptor, hydrophobic aliphatic and aromatic (π) contacts. We consider only nucleotide and dinucleotide binding sites with more than 3 surface exposed pseudocenters. Figure 1 presents examples of extruded nucleotide pairs and their protein dinucleotide binding sites.
Figure 1. Di-nucleotide binding sites.

(a) Parallel π-stacking interactions, as observed in the U1A spliceosomal protein (PDB: 1m5o, A6-C7 of U1 snRNA39). The surface of the protein binding site is represented as green dots. (b) The physico-chemical properties, termed pseudocenters, of the binding site in (a). Hydrogen bond donors are blue, acceptors - red, donors/acceptors - green, aliphatic - orange and aromatic - gray. (c) Non π-stacking dinucleotides of methyltransferase RumA (PDB: 2bh2, G1954-U1955). Although the rings participate in aromatic interactions, they are π-stacked with the protein and not with each other63.
The analysis below is performed on two datasets of protein-RNA complexes as well as a dataset of all RNA structures (see Table 1). The non-redundant datasets were constructed in the following way. Two RNA chains were considered redundant if they share more than 60% sequence identity35, 36. Two protein chains were considered redundant if they have more than 25% sequences identity34 or share a similar sub domain. The sub domain similarity was detected using the Pfam database, which contains a manually curated annotation of domains whose boundaries are also consistent with the SCOP20 structural data50. For each pair of redundant complexes we retained a single structure with the highest X-ray resolution. We used two options to define redundancy at the level of protein-RNA complexes. The first dataset, termed AND-set was constructed by removing those complexes that are redundant in both protein and RNA chains. It contained 154 complexes, with 288 non-redundant pairs of interacting protein-RNA chains. The second dataset, termed OR-set, was constructed by removing all the complexes that are redundant in at least one, either protein or RNA chain. It contained 92 structures with 95 interacting chains (see Table 1). The difference between the two datasets of protein-RNA complexes is in considering the structures that are similar in one interacting chain but are different in the other. For example, there are many examples of similar proteins that bind different RNA molecules. On the one hand, investigation of such interactions may provide insights into the binding specificity and can reveal the variability of the different protein binding platforms (see Figure 4 and Section 2.1). On the other hand, such cases may introduce bias into the overall statistics. Since each dataset has its limitations, validating the consistency of the results on both datasets, ensures the robustness of our methods and the correctness of the observations.
Table 1. Datasets statistics and the frequency of π-stacking.
The table presents the details of the constructed datasets as well as the statistics of π-stacking interactions between consecutive extruded nucleotides. Columns 1–4 present the dataset details and provide the numbers of redundant and non-redundant structures and chains. Column 5 details the number of pairs of consecutive extruded nucleotides, where only protein interacting nucleotides were considered in the dataset of protein-RNA complexes. Column 6 present the total numbers and the frequencies of parallel π-stacking interactions.
| Dataset | Structures total | Structures non-redundant | Chains non-redundant | Consecutive extruded pairs | π-stacked pairs |
|---|---|---|---|---|---|
| Protein-RNA AND-set | 322 | 154 | 288 | 375 | 124 (33%) |
| Protein-RNA OR-set | 322 | 92 | 95 | 156 | 53 (34%) |
| RNA structures | 1058 | 411 | 496 | 1564 | 538 (34%) |
Figure 4. Dinucleotide binding sites of RNA-binding domains.
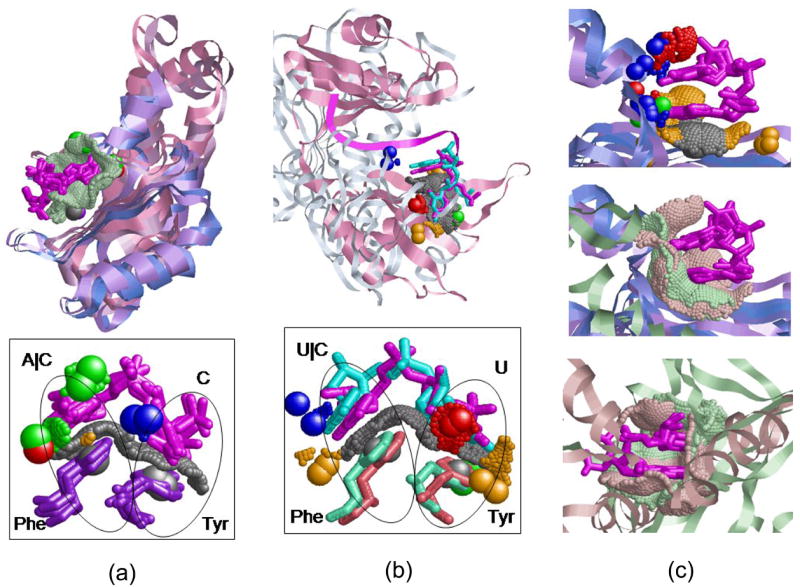
(a) Alignment between the six structures of cluster 94 in Table 2 according to the transformation calculated for the dinucleotide binding sites. The structures of RRMs are in variations of blue and the Rho termination factors are light and dark pink. The similar binding sites surfaces are green dots and the dinu-cleotides are magenta. The bottom figure details the common Tyr-Phe aromatic binding platform. The conserved Tyr and Phe amino acids (Y13-F56 of U1A/U2B and Y80-F64 of Rho) are represented as sticks and colored purple. The protein pseudocenters are as in Figure 1. (b) Alignment between the HuD protein (PDB:1fxl, pink) and the Rho termination factor (1pvo, monochrome) of cluster 23. The bottom figure presents the Tyr-Phe binding platform shared by the proteins. The conserved Tyr and Phe amino acids (Y128-F170 of HuD and Y80-F64 of Rho) are pink and green respectively. (c) Top: Alignment of the first four RRM members of cluster 124 (see Table 2), colored as in (a). Middle, the alignment of all the five cluster members, including the Aminoacyl-tRNA synthetase (PDB:1u0b, green). Bottom: Alignment of the binding sites of Poly(A)-binding protein (PDB:1cvj, green) and NSP3 homodimer (PDB:1knz, pink).
We analyzed the protein-RNA interactions in these datasets and observed that 80% of protein interacting extruded nucleotides have at least one RNA strand neighbor that interacts with the protein via its ring as well (see Supplementary Material). Moreover, many of them share the same binding cavity and 33–34% of these consecutive extruded nucleotides are π-stacked with each other (see Table 1 and Methods). When trying to estimate the sequence specificity of π-stacking, the only dinucleotide pair which was observed to have some tendency for π-stacking in all the datasets, was the AC pair (see Supplementary Material). This suggests that the local nucleotide geometries can not be predicted from the RNA sequence alone and nucleotide identities are not sufficient for the atomic level prediction of nucleotide interactions and spatial conformations. Consequently, we propose to utilize the protein binding patterns for the prediction of RNA nucleotide orientations and protein-RNA complexes. As illustrated in Figure 2 we classify the nucleotide and dinucleotide binding sites and use the common 3D patterns for the prediction of protein-RNA interactions.
Figure 2. The algorithmic framework.

The framework consists of two main stages. First, we classify all known nucleotide and dinucleotide binding sites and recognize the common types of the physico-chemical binding patterns (left flowchart). Then, we use these patterns for the prediction of dinucleotide binding sites and for the reconstruction of RNA fragments and protein-RNA complexes (right flowchart).
2.1 Classification of nucleotide and dinucleotide binding sites
We classify all the nucleotide and dinucleotide binding sites and create a non-redundant set of the 3D binding patterns, which describe their main types of interactions. We consider all the dinucleotide binding sites that accommodate pairs of consecutive extruded nucleotides. In addition, binding sites of single nucleotides that are not a part of such pairs are considered as single nucleotide binding sites.
We present a novel classification methodology which validates the cluster quality by multiple spatial binding site alignment. Specifically, we have developed a center-star classification algorithm, which creates clusters by iteratively adding binding sites in the order of their decreasing similarity (based on pairwise spatial alignments) and validates each new addition by the multiple spatial alignment among all current cluster members. If the multiple similarity, measured by the score of the common physico-chemical binding pattern, is lower than a predefined threshold (e.g. ≥ 30% of one of the binding sites) the new member is ignored and not added to the cluster. The main advantage of this approach is that we validate the spatial superimposition of the cluster members and assess the quality of the shared physico-chemical binding pattern.
We applied this methodology to two datasets of protein-RNA complexes and classified all the nucleotide and dinucleotide binding sites. As expected, many of the clusters obtained in the two classifications were very similar and the clusters in the OR-set were a subset of those in the AND-set. Figure 3(a) presents a histogram of the dinucleotide cluster sizes. As can be seen many of the binding sites were unique and were left as singleton clusters. The number of dinucleotide clusters with more than one member was 53 and 20 in the AND-set and OR-set respectively. Forty four percent of the significant clusters of the AND-set involve proteins with different sequences (less than 25% sequence identity and different Pfam annotations). The full details of all the clusters are provided in the Supplementary Material. Due to the low number of significant clusters of single nucleotide binding sites, which was 21 and 4 in the AND-set and OR-set classifications respectively, here we focus on the analysis of dinucleotide binding patterns. We distinguish between two types of clusters: (1) patterns formed by dinucleotide binding sites of proteins with similar structural folds; (2) patterns formed by dinucleotide binding sites of proteins with different structures. We start this section with a description of the clusters that involve members of the RNA-binding domain family which exhibit all of these types of similarities and illustrate the differences between our two datasets. Then, we present examples of additional similarities revealed by our classifications.
Figure 3.

(a) Clusters of dinucleotide binding sites. A histogram of the cluster sizes of the AND-set and OR-set classifications. The X-axis denotes the number of cluster members and the Y-axis is the number of clusters of this size. The top right pie chart shows the distribution of the cluster sizes of the AND-set. The distribution of clusters in the OR-set was very similar. (b) Success rates of the predictions of dinucleotide binding sites, measured by the leave-one-out tests. The X-axis denotes the number of the top ranking solutions considered in each prediction. The Y-axis is the overall success rate measured by the percentage of binding sites correctly predicted in all the leave-one-out tests performed with the AND-set and OR-set classifications.
2.1.1 The RNA-binding domains
The RNA recognition motif (RRM), also known as RNA-binding domain (RBD) or ribonucleoprotein domain (RNP) is considered to be one of the most abundant protein domains24. In spite of its overall structural simplicity this domain can recognize a wide variety of RNAs and can perform various biological functions. Our AND-set contained 9 complexes with RRMs of the following 6 types20, 38: (1) splicesomal U1A proteins (PDBs: 1urn:AP, 1m5o:CB, 1sj3:PR, 2nz4:AP); (2) splicing factor U2B” (PDB: 1a9n:BQ); (3) sex lethal protein, Sxl (PDB: 1b7f:AP); (4) HuD protein (PDB: 1fxl:AP) and (5) poly(A)-binding protein (PDB: 1cvj:AM); (6) Pre-mRNA splicing factor U2AF65 (PDB: 2g4b:AB). The clusters of the AND-set reveal the similarities and the differences of the various nucleotide binding sites which allow these proteins to achieve a range of required biological functions. As can be seen in Table 2, the created clusters divide our proteins into three main groups: (1) U1A and U2B” proteins that share more than 70% sequence identity and 5 similar dinucleotide binding sites; (2) HuD and Sxl, which share 50% sequence identity and 4 dinucleotide binding sites; (3) U2AF65 and poly(A) binding proteins, which are clear outliers. Alternatively, the OR-set contained only 2 complexes with U1A (1urn) and HuD (1fxl) proteins. These proteins have 19% sequence identity and the similarity of their dinucleotide binding sites is not high enough to be clustered together. Notably, our classifications are consistent with the available manual observations and comparisons which were previously performed for some of the complexes24, 25.
Table 2. Dinucleotide clusters that involve RNA-binding domain proteins.
The first column is the cluster number (according to the full list provided in the Supplementary Material). The second column specifies the maximal SCOP20, 62 distance between the cluster members. Proteins at distance 3 belong to the same SCOP super family, while proteins at distance 7 have totally different overall folds and no common structural annotation. The distance was measured according the pre-SCOP annotation when available62. Column 3 indicates whether the proteins have similar sequences, i.e. belong to the same Pfam family37 or have more than 25% sequence identity. Column 4 details whether all the cluster dinucleotide pairs are π-stacked (1) or non π-stacked (2). Column 5 indicates the number of binding sites in the cluster and column 6 provides the details of the binding site that initiated the cluster (PDB code, protein and RNA chains, nucleotides’ (nts) numbers according to the appearance on the RNA chain). Column 7 specifies the location of the considered dinucleotides according to the available literature and Column 8 details the cluster members.
| Cl. | Scop dist. | Sim. seq. | π type | Cl. size | Nts. pairs | Cluster center (pdb:chains:nts) | Location | Cluster members annotation |
|---|---|---|---|---|---|---|---|---|
| 97 | 1 | 1 | 1 | 3 | AU | 1sj3:PR:A49U50 | A1-U2 of U1 snRNA39 | U1A(1urn, 1m5o, 1sj3) |
| 120 | 3 | 1 | 2 | 3 | UU | 1a9n:BQ:U8U9 | U2-U3 of U1 snRNA39 | U1A(1urn,1sj3), U2B(1a9n) |
| 99 | 3 | 1 | 1 | 4 | UG | 1sj3:PR:U51G52 | U3-G4 of U1 snRNA39 | U1A(1sj3,1m5o,1urn), U2B(1a9n) |
| 100 | N/A | 1 | 2 | 5 | GC, UU | 1sj3:PR:G52C53 | G4-C5 of U1 snRNA39 | U1A(1sj3,1m5o,1urn), U2B(1a9n,2g4b) |
| 194 | 3 | 1 | 2 | 2 | GU, AA | 1b7f:AP:G4U5 | G4-U5 of 1b7f25 | Sxl(1b7f), Poly(A)(1cvj) |
| 195 | 3 | 1 | 2 | 2 | UU | 1b7fAP:U5U6 | U5-U6 of 1b7f25 | Sxl(1b7f), HuD(1fxl) |
| 25 | 3 | 1 | 2 | 2 | UU | 1fxl:AB:U3U4 | U6-U7 of 1b7f25 | Sxl(1b7f), HuD(1fxl) |
| 198 | 3 | 1 | 2 | 2 | UU | 1b7f:AP:U8U9 | U8-U9 of 1b7f25 | Sxl(1b7f), HuD(1fxl) |
| 157 | N/A | 0 | 2 | 2 | UU | 2g4b:AB:U5U6 | U9-U10 of 1b7f25 | Sxl(1b7f), U2B(2g4b) |
| 29 | 3 | 1 | 2 | 2 | UU | 1fxl:AB:U7U8 | U10-U11 of 1b7f25 | Sxl(1b7f), HuD(1fxl) |
| 28 | 6 | 0 | 2 | 2 | UU, UG | 1fxl:AB:U6U7 | U6-U7 of 1fxl, U210-G211 of 1sds | HuD (1fxl), Ribosomal protein L7ae (1sds) |
| 168 | 7 | 0 | 1 | 2 | AA, GA | 1cvj:AM:A4A5 | A4-A5 of 1cvj, G3-A4 of 1knz | Poly(A)(1cvj), NSP3 homodimer(1knz) |
| 124 | 7 | 0 | 1 | 5 | AG, AC, GC | 1a9n:BQ:A12G13 | A6-C7 of U1 snRNA39, G33-C34 of anticodon loop | U1A(1sj3,1m5o,1urn), U2B(1a9n), Aminoacyl-tRNA synthetase(1u0b) |
| 54 | 7 | 0 | 2 | 2 | CU, UG | 1n78:AC:C33U34 | U3-G4 of 1b7f25, C33-U34 of anti-codon loop | Sxl(1b7f), Glutamyl-tRNA synthetases(1n78) |
| 94 | 7 | 0 | 2 | 6 | CA, CC | 1m5o:CB:C40A41 | C5-A6 of U1 snRNA39, C1-C2 of 2a8v | U1A(1sj3,1m5o,1urn), U2B(1a9n), Rho termination factor(2a8v) |
| 23 | 7 | 0 | 2 | 2 | UU, UC | 1fxl:AB:U1U2 | U3-U4 of, U1-C2 of 1pvo | HuD(1fxl), Rho termination factor(1pvo) |
RRMs and proteins of other folds
In addition to the classification of the binding patterns within the family of RNA-binding proteins, the clusters detailed in Table 2 reveal novel similarities of RRMs to other structurally, functionally and evolutionary unrelated proteins. Below we describe several examples of such similarities, the first of which involves a well studied Tyr-Phe aromatic binding platform of RRMs.
RRMs are known to contain two highly conserved aromatic residues, Phe56 in RNP1 and Tyr13 in RNP2 (U1A numbering), which stack with the RNA bases39. Our clusters reveal the similarities and the differences of the spatial physico-chemical environments of these platforms. Specifically, while the platforms of the U1A and U2B” proteins were recognized to be similar, the binding pattern of the HuD protein was recognized to be different (see clusters 94, 23 in Table 2). Moreover, our AND-set contained three complexes of Rho termination factors which are different in their structure and sequence from RRMs and do not contain the required RNP sequence motifs. Notably, two of the structures were recognized to have a pattern similar to that of U1A and U2B” proteins, while the third structure was recognized to have an aromatic platform similar to the HuD protein. As can be seen in Figure 4, although the Rho termination factors are structurally different from RRMs, our method revealed an aromatic binding platform which binds dinucleotides in a manner very similar to RRMs. Furthermore, although the conformations of the aromatic residues are similar in all the complexes, the physico-chemical properties around them can be classified into two types of 3D patterns. These patterns, which are detailed in the Supplementary Material, are independent of the similarity of the overall sequences of these proteins. Most of these observations are lost in our OR-set which contained only a single Rho termination complex which was classified together with a single complex of U1A.
Figure 4 illustrates two additional examples of similarities of dinucleotide binding sites of RRMs to those of cysteinyl-tRNA synthetase and NSP3 homodimer (clusters 124 and 168 respectively). The first example shows the similarity between the dinucleotide binding sites of π-stacked A{C|G} pairs of U1A and U2B” to the binding site of a π-stacked anticodon pair G33–C34 of cysteinyl-tRNA synthetase (PDB: 1u0b). The second example, reveals the similarity between the poly(A)-binding protein and the NSP3 homodimer (PDB: 1knz). In both examples, in spite of the structural differences between the proteins, the binding sites have similar surfaces and shapes and bind the π-stacked nucleotide pairs in exactly the same orientation. A very similar situation is observed in the rest of the clusters of this type, which are detailed in Table 2.
2.1.2 Similar structures, similar dinucleotide binding sites
Similar binding patterns formed by structurally similar proteins are expected and are not surprising. This type of similarity is usually well illustrated in the biological literature, which allows us to verify the correctness of the recognized physico-chemical patterns. In addition, the correct superimposition of the overall structures by the transformations calculated for the dinucleotide binding sites allows us to validate the correctness of the alignment. Table 3 details all the recognized clusters of this type. Below we describe several clusters that provide some insights into the RNA sequence specificity and reveal the repetitive nature of certain binding patterns.
Table 3. Similar structures, similar dinucleotide binding sites.
Clusters comprised of dinucleotide binding sites of structurally similar proteins. The columns are the same as in Table 2.
| Num. | Scop dist. | Sim. seq. | π type | Cl. size | Matched Nts. | Cluster center (pdb:chains:nts) | Location | Protein annotation |
|---|---|---|---|---|---|---|---|---|
| 812 | 0 | 1 | 2 | 3 | UGU, UAU | 1m8x:AC:U2G3 | Pumilio repeats R6–8, R2–4, R4–6 43 | Pumilio-homology domain (1m8x) |
| 159 | 1 | 1 | 2 | 6 | UA, CA, AA | 2izn:AR:U9A10 | 4nt-loop: -5, -442 | RNA bacteriophage capsid proteins (2izn, 1zdj, 7msf, 6msf, 2bs0, 2b2d) |
| 175 | 1 | 1 | 1 | 2 | AA | 1g1x:BE:A13A14 | A728-A729, helix 23a, GAAG tetraloop 64 | Ribosomal protein S15 (1g1xB, 2j02O) |
| 178, 2801 | 1 | 1 | 1 | 2 | GCCG | 2j02:RA:G684C685 | G718-G720 on helix 23a 64 | Ribosomal protein S18 (2j02R, 1g1xC) |
| 299 | 1 | 1 | 2 | 2 | AA | 1m90CA:A862A863 | A781-A782 of 2j01 | Ribosomal protein L2 (1m90C, 2j01D) |
| 242 | 1 | 0 | 2 | 2 | AG, AU | 2j01:FA:A293G294 | A327-U328 of 1m90E | Ribosomal protein L4 (2j01F, 1m90E) |
| 78 | 1 | 1 | 2 | 3 | GC, UC | 1yvp:AG:C2C3 | U11-C12 of 2i91 | 60-kda SS-aARo ribonucleoprotein (1yvp, 2i91) |
| 184, 137 | 1 | 1 | 1 | 2 | GCC | 1il2:AC:G64C65 | 3′ GCCA65 | Aspartyl-tRNA synthetases (1c0a, 1il2) |
| 134 | 2 | 1 | 2 | 3 | UC | 1c0a:AB:U31C32 | Anticodon loop U635-C636 65 | Aspartyl-tRNA synthetases (1c0a, 1il2, 1asy) |
| 182 | 2 | 0 | 2 | 2 | GU | 1il2:AC:G30U31 | G30-U31 at loop base | Aspartyl-tRNA synthetases (1il2, 1asy) |
| 208 | 2 | 1 | 2 | 2 | UA, AA | 1i6u:AC:U25A26 | U640-A64166 | Ribosomal protein S8 (1i6u, 1s03) |
| 58, 105 | 3 | 0 | 1,2 | 2 | CCA | 1n78:AC:C72-A74 | 3′ CCA | Glutamyl (1n78) and Glutaminyl (1qtq) tRNA synthetases |
| 164,412 | N/A | 0 | 2 | 3 | UCAC, CCCU | 2anr:AB:U12C13 | protein KH motif | KH-domain (2py9, 1ec6, 2anr) |
| 202 | N/A | 1 | 2 | 4 | CA, AA | 2ozb:AC:A10A11 | Rna A29-A30 of 1e7k | L30e/L7ae Ribosomal proteins (1e7k, 1m90H, 2hvyD, 2ozbA) |
| 21 | 3 | 1 | 2 | 6 | GU, AU | 1sds:AD:G9U10 | Rna A30-A31 of 1e7k | L30e/L7ae Ribosomal proteins (2ozbA, 1e7k, 1m90H, 1sds, 2hvy, 1rlg) |
Nucleotide sequence specificity
MS2 RNA hairpin coat-protein complexes, have been widely used as a model system for studying RNA sequence specificity40, 41. Our AND-set contained 6 complexes of RNA bacteriophage capsid proteins, most of which were determined as part of these studies. These studies have revealed that the main driving force for the complex formation is the π-stacking interaction between the -5 base and the conserved tyrosine side chain. Our clusters support this finding. First, they reveal that the only extruded dinucleotides which have a sufficient contact with the protein are nucleotide bases -4 and -541 (see Table 3 cluster 215). Second, they recognize the physico-chemical binding pattern common to all binding sites of this dinucleotide pair (see Figure 5(a)). They clearly show the conservation of the critical tyrosine side chain (Tyr85, PDB: 2izn), which can tolerate any nucleotide sequence mutations (U|G|A|C)42. In addition, the specific pattern recognized for the binding site of adenosine, can explain the specificity of this position to this nucleotide type42.
Figure 5. Dinucleotide binding sites of proteins with similar structures.

(a) Alignment between 6 dinucleotide binding sites of MS2 RNA hairpin coat-proteins (cluster 159, Table 3). The left binding site can tolerate any nucleotide sequence, due to the conservation of its Tyr residue, represented by two pseudocenters: donor/acceptor (green) and aromatic (black). The pseudocenters which describe the adenine pattern are colored as in Figure 1. (b) Alignment between three repetitive binding sites of Pumilio proteins (see cluster 81–82, Table 3). The proteins of the repeats R2–4, R4–6 and R6–8 are cyan, purple and light blue. The surfaces of the binding sites are represented by dots, colored by the proteins.
Repetitive Protein Patterns
Our datasets contained a structure of a Pumilio protein (PDB: 1m8x), that regulates mRNA expression43. Interestingly, our classification revealed similar patterns of interactions that appear in three different regions of this protein (see clusters 81–82 in Table 3). This is consistent with the previous, crystallographic studies of this protein which manually analyzed the interactions of the regions, termed repeats 2–4, 4–6 and 6–8. The contribution of our method to these previous observations is the recognition of conservation of the spatial physico-chemical binding patterns of these regions that bind RNA nucleotides in a similar manner (see Figure 5(b) and Supplementary Materials).
K homology (KH) motifs
The K homology (KH) is a widespread RNA-binding sequence motif, which spans about 70 residues with a characteristic pattern of an invariant Gly-X-X-Gly segment44. Our datasets contained 3 structures of proteins with KH-motifs: (1) Nova-1 (PDB: 2anrA); (2) Poly(rC)-binding protein 2 (PDB: 2py9A); (3) Neuro-oncological ventral antigen 2, Nova-2 (PDB: 1ec6A). Since the overall sequence identity among these three proteins is less than 20%, they appeared in both AND-set and OR-set and formed exactly the same clusters in the two classifications. These clusters, which are detailed in Table 4, recognized the similarity between 3 consecutive dinucleotide binding sites. They reveal the remarkable conservation of the binding patterns of the four corresponding consecutive nucleotides UCAC (2anr,1ec6) and CCCU (2py9). Figure 6 shows the surfaces of these binding sites which have almost identical physico-chemical properties and shapes. The common pattern, detailed in Supplementary Materials, is consistent with the previous manual analysis of the separate crystal structures19, 44. However, no previous studies have compared the spatial arrangement of the physico-chemical properties up to the level of surface points that they create.
Table 4. Different structures, similar dinucleotide binding sites.
Clusters comprised of similar dinucleotide binding sites of proteins with different overall folds. The columns are the same as in Table 2 except the column which indicates whether the proteins belong to the same Pfam family. It was removed since none of the clusters was comprised of proteins with more than 25% sequence identity or similar Pfam annotation.
| Num. | π type | Cl. size | Nts pairs | Cl. center (pdb:chains:nts) | Location | Cluster members annotation |
|---|---|---|---|---|---|---|
| 103 | 2 | 2 | CU, AA | 1qtq:AB:C32U33 | -2,-3 AA at the PAP binding site of 2q66 | Glutaminyl-tRNA synthetase (1qtq) and tRNA-guanine transglycosylase (2q66) |
| 55 | 3 | 3 | UC, AG | 1n78:AC:U34C35 | UC of anticodon loop, A11-G12 of 2gzt | Glutamyl-tRNA synthetase (1n78) and Hut operon regulatory protein (1wpu, 2gzt) |
| 354 | 2 | 2 | CU, CA | 1q2r:AE:C8U9 | 3′ CCA | tRNA-guanine transglycosylase (1q2r) and Aminoacyl-tRNA synthetases (1u0b) |
| 59 | 2 | 2 | AG, AU | 1jbr:AC:A16G17 | A700-U701 of 1m90 | Restrictocin (1jbr) and Ribosomal proteins L18e (1m90P) |
| 12 | 2 | 3 | UA, GA | 2gzt:AD:U10A11 | U641-A-642 of 1j02 | Hut operon regulatory protein (2gzt, 1wpu), Ribosomal protein S8 (2j02:H) |
| 235 | 2 | 2 | GG | 2j01:7A:G116G117 | G147-G148 of 1jid | Ribosomal protein L34 (2j01), SRP19 (1jid) |
| 61 | 1 | 2 | UC, AC | 2bh2:AC:U8C9 | 5′ loop U1940-C1941 stack63, A11-C12 of 1hq1 | rRNA methyltransferase (2bh2), Signal sequence binding protein (1hq1) |
| 67 | 2 | 2 | GU, AG | 2bh2:AC:G22U23 | hairpin recognition G1954-U195563, A35-G36 of 2du3 | rRNA methyltransferase (2bh2), O-phosphoseryl-tRNA synthetase (2du3) |
| 76 | 2 | 2 | CG, CA | 1yvp:AC:C7G8 | 3′ CCA of t-RNA | 60-kda SS-aARo ribonucleoprotein (1yvp), Aspartyl-tRNA synthetases (1il2) |
| 52 | 3 | 3 | CG, AA | 1r3e:AC:C7G8 | part of a UCG flipped out triplet67 | Pseudouridine synthase II TruB (1r3e,1k8w) and Ribonuclease II (2ix1) |
Figure 6.

(a) Alignment of 3 binding sites of proteins with KH-motifs, detailed in clusters 164, 41, 42. The nucleotides, UCAC (2anr,1ec6) and CCCU (2py9) are represented as sticks, colored cpk. Surface patches with exactly the same properties and shapes (up to 1Å distance deviation) are represented as dots colored as in Figure 1. (b) Alignment between the 3 binding sites of cluster 12: Hut operon regulatory proteins (PDB: 2gzt, 1wpu, blue) and Ribosomal protein S8 (PDB: 2j02H, pink). The corresponding nucleotides GA, UA and GA are green, yellow and orange. (c) Alignment of the AG dinucleotide binding sites of the Hut proteins (blue and green) to the UC binding site of the anticodon loop of Glutamyl-tRNA synthetases (PDB:1n78, pink).
2.1.3 Different structures, similar dinucleotide binding sites
As illustrated above, similarity of the sequence patterns can lead to the similarity of the dinucleotide binding sites. Since our methodology does not assume the similarity of the protein sequences or structures, many of our clusters, detailed in Table 4, reveal similar dinucleotide binding sites of proteins that do not share any known motifs and are not evolutionarily related. Figure 6 presents two examples of two consecutive dinucleotide binding sites of Hut operon regulatory proteins (HutP, PDBs: 1wpu, 2gzt). The first binding site, formed with the pairs G3-A4 (1wpu) and U10-A11 (2gzt) was aligned to a binding site of the Ribosomal protein S8 (2j02). The second binding site of the pairs A4-G5 (1wpu) and A11-G12 (2gzt), was aligned to the UC binding site of the anticodon loop of Glutamyl-tRNA synthetase (1n78). As can be seen, in spite of the structural differences of the aligned proteins the surfaces of the binding sites are very similar and they bind the RNA nucleotides in very similar conformations.
To summarize, similarities of this type are very interesting, unexpected and can not be recognized by other methods. However, since many of the aligned proteins are functionally unrelated we can not assess the biological meaning of these similarities. Currently, we can only speculate that certain binding patterns that are favorable for RNA recognition are reused in different protein regions. Consequently, the main contribution of this type of clusters is in their application for the prediction of RNA binding sites which we show below.
2.2 Prediction of RNA binding sites and structure
Given a target protein structure, we aim to recognize the binding sites of the extruded nucleotides and to construct the RNA fragments that can bind to them, predicting the structure of the RNA strand and of the protein-RNA complex. Figure 2 illustrates the flow of our classification and prediction processes. Specifically, given the clusters of binding sites described in the previous section, we define the 3D consensus binding patterns by the physico-chemical properties shared by all cluster members (see Methods). Given a target protein structure not used in the classification we search its surface for the presence of any of these 3D consensus patterns, predicting its dinucleotide binding sites. Using the nucleotide orientations observed in the consensus patterns allows us to predict the structure of RNA strands and protein-RNA complexes.
2.2.1 Prediction of RNA binding sites
Here, we use the created clusters to predict RNA binding sites that accommodate unpaired extruded nucleotides. Specifically, given a target protein structure not used for the classification, we search its surface for regions similar to the created 3D consensus binding patterns. These regions are predicted to serve as binding sites. Since the created 3D patterns provide a non-redundant description of the main types of interaction, the prediction procedure is extremely efficient and takes only several minutes on a standard PC. It must be noted, that currently, we do not aim to predict whether a protein can bind RNA, rather given an unbound RNA binding protein our goal is to predict its binding sites and their modes of interaction. Due to a low number of single nucleotide clusters, we evaluate our predictions of dinucleotide binding sites. We perform leave-one-out tests on all the structures that participated in the above described clusters with more than one member. For each left out complex, we repeat the classification procedure without its structure, and use the obtained clusters to define a new set of 3D consensus binding patterns. Then, we search the surface of the left out protein for a presence of the constructed 3D patterns. All of these are searched by a single algorithm (RnaPred, see Methods) which recognizes the top ranking patterns that are similar to some protein regions. For each dinucleotide binding site of each left out protein we check whether it was correctly predicted by this procedure.
Since most of the proteins have several dinucleotide binding sites, the output of the RnaPred method describes a set of different protein regions that resemble some of the constructed 3D patterns. Many of these are correct predictions of different dinucleotide binding sites of the same protein. The ranking of these solutions is not straightforward45. For example, if a certain protein has 10 dinucleotide binding sites, our goal is to have all of them listed as different solutions in the output of our method. In our current implementation, binding sites aligned to larger patterns will usually be the top ranking and smaller alignments will receive a lower rank. Consequently, to calculate the rate of the correct predictions we need to consider a certain number of top ranking solutions. Figure 3(b) presents our prediction success rates as the function of the number of considered top ranking solutions. Only binding sites that are correctly predicted within the given number of top ranking solutions are considered to be a success (see Methods). The percentage of binding sites that were successfully predicted in all the leave-one-out tests is the success rate of our methodology. As can be seen, when we consider 100 top ranking solutions, the success rates achieved with the AND-set and OR-set classifications are 91% and 86% respectively. These rates are significant since the top one hundred solutions represent approximately 0.01% percent of all potential alignments. However, the manual investigation of such a large number of solutions is not feasible. On the other hand, since some proteins have 10 dinucleotide binding sites, we can not consider less than 10 solution. Consequently, we suggest that 20 top ranking solutions provide a reasonable cut o_ to measure the prediction quality. Using this threshold, the success rates of the predictions obtained with our AND-set and OR-set classifications are 75% and 70% respectively. Thirty two percent of the correct predictions made with the AND-set classification were based on proteins with different sequences (less than 25% identity and no common Pfam domain). Notice, that our results, in addition to pointing to the specific amino acids involved in the interactions, predict the spatial orientation of the RNA nucleotides in the protein binding site. Consequently, we expect that the combination of such predictions will allow the reconstruction of protein-RNA complexes with an unknown structure. Below, we detail our preliminary attempts in achieving this goal.
2.2.2 Prediction of RNA strands structure
Given a protein structure, we would like to be able to predict which RNA fragments it can bind and what is the structure of the resulting protein-RNA complex. This is a very ambitious goal and currently we only show some initial steps in achieving it.
Existing tools are unable to provide a good solution to this problem for several reasons. First, we aim to solve this problem without any assumption or knowledge of the structure of the RNA molecule. Due to the limited number of existing RNA structures, this is an important requirement. Unfortunately, it prevents using standard methods like docking for its solution. Currently, the most applicable approach is the superimposition of the given protein upon the protein-RNA complex of its closest homologue. This provides the prediction of the complex between the input protein and the RNA molecule bound to the homologue. However, this approach has several limitations. First, similar overall sequences and folds do not always lead to similar nucleotide binding modes. Second, the superimposition of proteins done by their backbone atoms often misaligns the RNA molecules and the specific nucleotide binding sites of interest. Our methodology improves these points in the following ways. First, we do not require the existence of a homologous protein-RNA complex. Second, by looking for the similarity of the physico-chemical patterns in the binding sites, we focus on the protein information which indeed leads to the similarity in binding. Moreover, we superimpose the proteins according to the transformations calculated for their binding sites which usually optimize the similarity of the RNA nucleotide orientations.
Here, we extend the RnaPred algorithm to the following scheme. Given a protein structure, we first recognize all regions that are similar to the above described 3D consensus patterns. Then, we superimpose the matched 3D patterns upon our target structure and consider the RNA dinucleotides that are bound to these patterns. We consider all the solutions and check whether the nucleotides superimposed on neighboring regions can be combined to form a continuous RNA fragment. The longest RNA fragment with the highest similarity score of the alignment between the protein binding sites and the selected 3D patterns is the top ranking solution. Since the constructed RNA fragment is comprised of dinucleotides taken from 3D consensus patterns based on binding sites of totally different proteins, it can be different from any RNA fragment of known sequence and structure. This allows us to make unique predictions that can not be achieved by other methods.
To evaluate the performance of this method, we have performed leave-one-out tests similar to those described in the previous section. As before, each time we have left out one protein structure, re-clustered and created a new set of 3D consensus patterns that do not assume any knowledge of the left out structure. Then, this structure was searched for the highest scoring set of regions similar to the constructed 3D consensus patterns that allow us to create the longest continuous RNA fragment. The average length of the predicted fragment was 5 and the average running time on a standard PC was 3.5 minutes (AMD Opteron 242, 1593MHz). When we calculated the RMSD between the predicted RNA fragments and the real fragments bound to the left out structures, in 23% of the cases it was less than 5Å, and in 13% of the cases it was even less than 1 Å. In most of the remaining cases, although we have reconstructed some sub-fragments, we have added false positive predictions which pointed to regions that are not in interaction in the given complex.
Figure 7 presents two examples which illustrate our success and limitations. In the first example, we reconstruct part of the RNA hairpin which interacts with the Nova-1 KH domain (PDB: 2anr). The protein-RNA interaction of this complex involves 5 consecutive RNA nucleotides, 4 of which were described above in the clusters 41–42, and 164 and were presented in Figure 6(a). Since these binding sites were recognized to contain patterns similar to other proteins, we had enough information for the prediction. Specifically, when the structure of Nova-1 was left out, both the AND-set and the OR-set contained three clusters formed by the binding sites of Nova-2 and Poly(rC)-binding protein 2. The 3D patterns that represent these clusters were correctly mapped to the structure of Nova-1 and the nucleotides bound to these patterns allowed predicting the structures of the interacting RNA fragment consisting of 4 nucleotides. The fifth nucleotide was not predicted because its binding pattern was unique to Nova-1 and had no similar binding sites.
Figure 7. Structure prediction of RNA fragments.
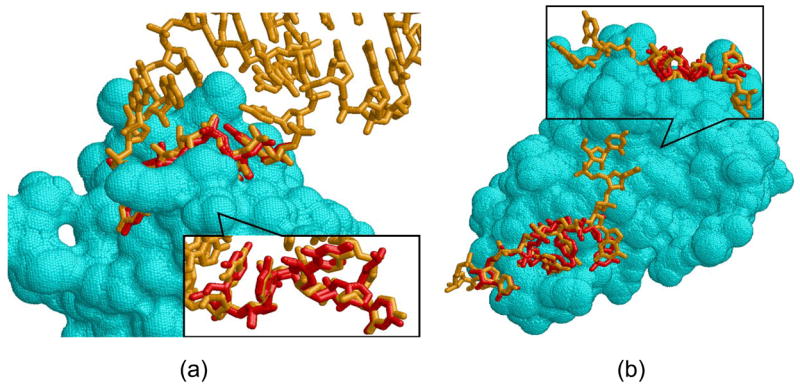
(a) Reconstruction of part of the RNA hairpin which interacts with Nova-1 KH domain (PDB:2anr). The predicted nucleotides are red and the native are orange. (b) Reconstruction of the RNA fragment of HutP antitermination complex (PDB:1wpu). The predicted and the native nucleotide fragments are red and orange sticks respectively. The opposite view in the upper figure shows the non interacting U nucleotide, not predicted by our method.
Figure 7(b) presents another example in which we reconstruct part of the HutP antitermination complex (PDB: 1wpu). The total length of the RNA strand in this complex is 7 nucleotides and it interacts with the protein through 4 dinucleotide binding sites. When we applied our prediction method and searched the surface of the Hut protein with the patterns of the AND-set classification, we have correctly predicted 3 of these dinucleotide binding sites. Only one binding site was predicted based on a pattern from another Hut protein (PDB: 2gzt) and two other were predicted based on patterns from Aspartyl-tRNA synthetases (PDBs: 1il2, 1c0a). Interestingly, the RNA nucleotides bound to these patterns provided a better backbone connectivity than those bound to the above described Glutamyl-tRNA synthetase and Ribosomal protein S8. In spite of the fact that the prediction was based on totally unrelated proteins, the patterns were correctly mapped to the query protein and the structure of the RNA fragment was correctly predicted with an overall RMSD of 0.9Å from the native. However, the structure of two 5′UU nucleotides was not predicted by our method. The first 5′U was ignored due to some missing atoms in its structure. Since the second U nucleotide has no interaction with the protein, its binding site and ring orientation could not be predicted by our method (see Figure 7(a) top). Interestingly, this nucleotide is also more flexible than the rest and its prediction is more difficult. Since our OR-dataset did not contain the additional structure of the Hut protein as well as the structures of both Aspartyl-tRNA synthetases, the prediction based on this classification consisted of only one correctly predicted dinucleotide pair with an RMSD 1.9Å from the native. This was predicted based on the pattern from Glutamyl-tRNA synthetase, which could not be connected to the pattern from the Ribosomal protein S8 due to backbone connectivity constraints violation. This example shows that due to the sensitivity of our method to local nucleotide flexibilities, we need a variety of structures to obtain a diversity of 3D patterns required for the prediction.
2.3 Drug design applications
There are two main types of drugs that can be developed to prevent the formation of protein-RNA interactions. The first type are the most common small molecule drugs, which are bioavailable when administered orally. The second type are drugs based on short strands of RNA oligonucleotides. These ligands, known as aptamers are selected for their ability to bind proteins with both high affinity and high specificity46. Below we present drug discovery applications and show how our methodology can contribute to the development of both type of drugs.
2.3.1 Discovery of small molecule drugs
We have observed that the average surface area of protein binding sites that accommodate dinucleotide pairs is 145 Å 2 if the pairs are π-stacked and 210 Å 2 otherwise. Since optimal drug molecules were proposed to have a surface area smaller than 140 Å 2 due to bioavailability reasons47, the dinucleotide binding sites have a high potential to accommodate drugs and serve as drug targets. To provide suggestions of potential ligands and ligand fragments that can target dinucleotide binding sites, we have searched the constructed 3D consensus patterns against a database of all known small molecule binding sites (see Methods). The top ranking solutions of this search application are the binding sites with properties similar to dinucleotide binding sites. The drug leads and the substrates that are bound to them provide ideas for small molecules that can bind to the dinucleotide binding sites. Figure 8(a) presents one example obtained in these searches. In this solution the binding pattern defined by cluster 81 in Table 3 was aligned to the binding site of Thrombin bound to an inhibitor (PDB: 1nzq, rank 9). The 3D binding pattern was represented by the pseudocenters from the G14–U15 binding site on the repeats 2–3 of the Pumilio protein (PDB: 1m8x). As can be seen, the inhibitor molecule has a volume similar to the GU nucleotide pair. Moreover, both the inhibitor and the G nucleotide form π-stacking interactions with the Tyr side-chain (Tyr-60, 1nzq and Tyr-1123, 1m8x), which has the same spatial arrangement in both binding sites.
Figure 8. Searching the database of drug-like binding sites.
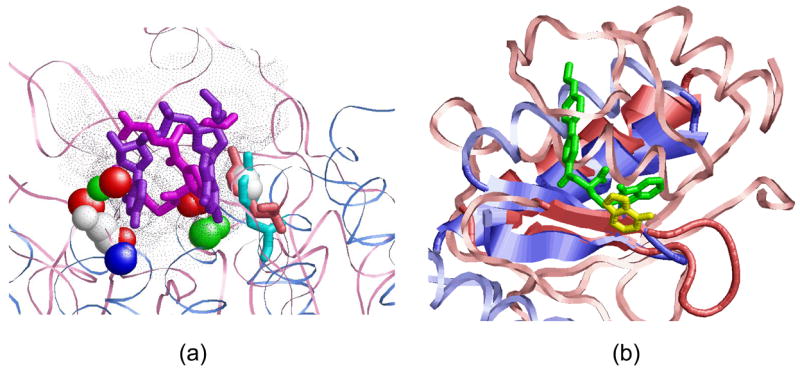
(a) Alignment of the dinucleotide binding pattern of cluster 81, which was found to be similar to the binding site of Thrombin in complex with an inhibitor (PDB:1nzq, rank 9) (b) An adenine binding site (A37, yellow) of Cysteinyl-tRNA synthetase (1u0b, light blue), whose 3D pattern was found to be similar to the binding site of Human macrophage elastase (1ros, pink) bound to its inhibitor (green). The aligned proteins have similar binding sites and similar secondary structure elements (helix and 2 strands), which suggest the potential similarity in ligand binding.
2.3.2 Aptamer design and optimization
Given a protein structure, our RnaPred method makes knowledged-based predictions and can suggest previously unknown RNA sequences and structures that can bind to it. Consequently, it may be used to suggest and optimize the RNA sequences in the aptamer design process. For example, one way to improve the binding affinity and/or selectivity of aptamers is to optimize the single extruded nucleotides, which are unpaired and are not part of a protein interacting dinucleotide pair. To obtain ideas for the chemical groups and scaffolds that can be used for such modification, we have searched the above described database of drug-like binding sites for those that are similar to the nucleotide binding sites. Figure 8(b) presents one example of the similarity between the 3D patten of an adenine nucleotide (A37) binding site of Cysteinyl-tRNA synthetase (PDB: 1u0b) to the binding site of Human macrophage elastase (MMP-12, PDB: 1ros) bound to its inhibitor. Since the binding regions of two proteins have similar physico-chemical properties and secondary structure elements (helix and 2 strands), the inhibitors developed for the MMP-12 can provide useful ideas for the chemical groups that can be used to substitute and optimize the adenine nucleotide which binds to Cysteinyl-tRNA synthetase. The predictions by a computational method provide a set of suggestions, which require further validation and optimization.
3 Summary and conclusions
Motivated by the important role of extruded non-paired RNA nucleotides in protein-RNA recognition, we have investigated their local geometries and interactions. We have observed that in most cases of protein-nucleotide interactions, there are several consecutive RNA nucleotides that are not involved in RNA base pairing. Since the nucleotide identities are not indicative of their spatial geometry, we consider the protein pockets that accommodate them. We observed that many of the consecutive nucleotide pairs share the same binding cavity and interact with each other. Consequently, we suggest that the protein binding patterns of such nucleotide pairs provide a more correct representation of their interactions.
We proposed a novel algorithmic framework which starts with the classification of all known nucleotide and dinucleotide binding sites according to their spatial physico-chemical patterns. These clusters, define a set of 3D consensus patterns, which provide a non-redundant representation of the main types of extruded nucleotide interactions. We show that these patterns can be efficiently used for the prediction of binding sites and RNA fragments. Obviously, the proposed framework is just a starting point and each of its stages can be further enhanced and improved. The classification methodology, which has the advantage of the spatial validation of the created patterns, shares the disadvantages of the regular center star clustering and is sensitive to the selected star centers and the order of traversal. The created 3D consensus patterns, which were shown to be extremely useful, do not contain the information about the variation of the spatial patterns of the cluster members, whose description is not straightforward. Selection of the shared pattern coordinates from a single structure could further influence the results. Currently, we do not predict the interactions formed with the RNA backbone, which are often represented by smaller and more flexible physico-chemical binding patterns. Nonetheless, the results of this paper indicate that it is possible to predict the interactions and the structure of fragments of single-stranded RNA bases. We intend to use the methodology presented here as the first part in a two stage scheme for the prediction of complete protein-interacting RNA strands. The results of this paper allow the prediction of binding sites and interactions formed with the nucleotide bases. Modeling the short RNA sub-fragments between the predicted regions is expected to allow the challenging reconstruction of complete RNA fragments.
In addition to being an important milestone towards achieving the ultimate RNA-protein structure prediction goal, our results provided several important insights. First, the presented classification reveals novel and surprising similarities between dinucleotide binding sites formed by proteins with different overall sequences, folds and functions. Our results suggest that certain physico-chemical patterns may be reused during the evolution in different protein regions that are important for RNA recognition. Second, we have presented a framework which allows a successful prediction of dinucleotide binding sites as well as recognition of ligands and ligand fragments that can target them. We hope that this will be useful in the design of aptamer and small molecule drugs that interfere with protein-RNA interactions.
4 Methods
Dataset construction and analysis
The structures of RNA and protein-RNA complexes were retrieved from the NDB database48, December 2007, and contained 1031 and 322 structures respectively. The dataset of protein-RNA complexes contained only structures with resolution 3Å and better, while the dataset of all RNA structures contained NMR structures as well. Sequence redundancy was removed using the ClustalW software49. Only the standard, unmodified, RNA nucleotides A,G,C,U, I were considered. Their local base pairing was recognized using the 3DNA software 51. We define a pair of consecutive extruded nucleotides as π-stacking, if the corresponding nucleotides’ planes are either parallel or perpendicular and have an angle of 180 ± 20° or 90 ± 20° respectively. In addition, we require that the distance between nucleotide ring centroids is less than 7Å52. In order to prevent the influence of missing strands which are part of a double helix, sequences of more than 5 consecutive π-stacked nucleotides were not considered in the statistics.
Alignment of nucleotide and dinucleotide binding sites
As described above, we consider the protein binding sites represented by their surfaces and surface exposed pseudocenters. Figure 9(a), provides an example of pseudocenters extracted from the protein amino acids. Each pseudocenter is assigned such attributes as charge, normal vectors of the surface direction, ring plane orientation as well as surface patch size and curvature53, 54. Since it was shown that single nucleotides can bind in alternative modes even to the same protein binding site55, the alignment of their binding sites was performed, using our previously developed, MultiBind method53. This method allows the recognition of the maximal physico-chemical pattern common to the input set of binding sites, without using any information regarding the corresponding binding partners.
Figure 9.
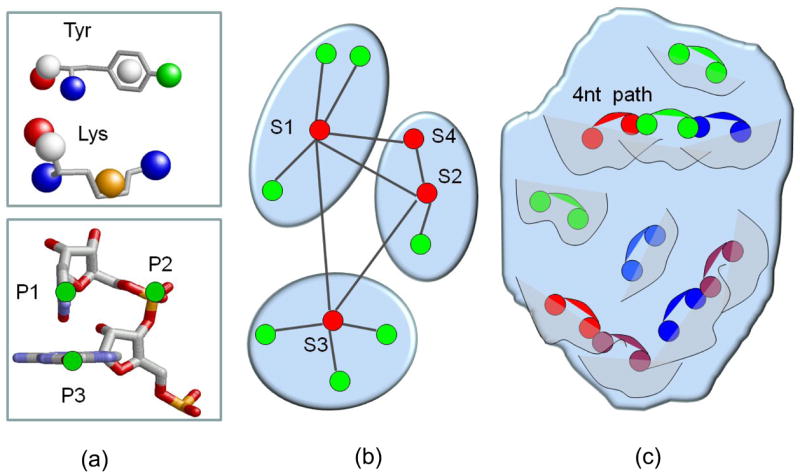
(a) Representation of the protein and RNA molecules. Top: An example of pseudocenters extracted from the side chains and the backbone of Lysine and Tyrosine amino acids. The pseudocenters are represented as balls colored as in Figure 1. Bottom: Representation of a dinucleotide pair by three points: two nucleotide rings centroids (P1 and P3) and the backbone phosphate atom between them (P2). (b) Center star classification algorithm. The nodes s1–s4 are enumerated in the order of their start weights and traversal. The edges of s1 to s2–s4 nodes failed to fulfill the multiple alignment requirements and were not added to the cluster of s1. The algorithm proceeded to check the star of s2, where s4 fulfilled the multiple alignment requirement and was added to the cluster. (c) The RnaPred algorithms aligns the 3D consensus patterns to some regions (represented by curves) of the complete query protein. We select the highest scoring set of alignments of 3D patterns to neighboring regions so that their bound dinucleotides (pairs of connected balls) can be combined to form the longest continuous RNA fragment (of 4 nucleotides in this example).
The dinucleotide binding sites extracted from protein-RNA complexes contain additional information about the spatial orientation of its two nucleotides, which we aim to predict. Consequently, it is essential that the alignment method requires the similarity of the nucleotide geometries in the aligned binding sites. To fulfil this requirement, we have developed a new method, RnaBind, which aligns between dinucleotide binding sites and utilizes the nucleotide orientation for the construction of 3D transformations that superimpose the input binding sites. Specifically, each dinucleotide pair is represented by the two centroids of the corresponding nucleotide rings and the phosphate atom between them (see Figure 9(a)). Then we apply the Least-Squares Fitting method56 to calculate the transformation that provides the best alignment of such representative triplets extracted from the input binding sites. Once the binding sites are superimposed in 3D space we apply maximum weight match in a weighted bipartite graph57 to determine the 1:1 correspondences between the matched pseudocenters of the input binding sites. The score of the alignment is the sum of similarity scores of the matched pseudocenters. These are measured by a scoring function that compares properties like spatial proximity (after the superimposition), charge and surface curvature. In addition, we score the overlap of the corresponding superimposed physico-chemical surfaces (surface points within the distance of 1Å) as well as the similarity of the corresponding aromatic ring plane orientations53, 54.
Multiple center star clustering
The standard clustering methods such as UPGMA or k-means 58 provide a general methodology to group any elements based on their pair-wise relations. Obviously, some complex elements that may be similar between pairs, may not be similar as a whole group. For the clustering of dinucleotide binding sites we are interested in computing clusters of binding sites that are similar as a whole group, i.e. finding a group of binding sites that share a significant 3D consensus pattern. Therefore, we developed the following new clustering procedure.
Similar to most existing methods, we start with performing all-against all pairwise alignments between objects of interest, which in our case are nucleotide and dinucleotide binding sites. Then, we use the calculated pairwise scores to create a graph in which the binding sites are the nodes. Edges are created only between nodes, which have more than 30% similarity. This is measured relative to the score of a binding site aligned to itself. Then, for each node, we consider the “star” that is created by its edges and determine the star weight by the sum of scores of these edges. We sort all the nodes which represent our binding sites, in the decreasing order of the weight of the stars that they create. In addition, we sort the edges of each star in the decreasing order of their score. Then, we traverse all the stars and all their edges in this order and check which members of a star can be classified together in the same cluster. Here, we impose a strict requirement of the similarity of the spatial patterns of the cluster members. This is measured by the score of the multiple spatial alignment which is performed between the binding sites of each star. Specifically, we start with a node that creates the highest scoring star and go over its edges in the decreasing order of their score. We start with the highest scoring edge and perform the alignment between its two nodes. Then, we add the second edge and perform a multiple alignment between three nodes: the star center and the two nodes of its two highest scoring edges. If the score of the core of this multiple alignment is above 30% of the score of each node, this triplet is defined as a cluster. Otherwise, the last node that was just added in this iteration is removed from the cluster. It will be considered later according to its other edges. As to the current star, we proceed going over the rest of its edges and add to the cluster only those nodes whose multiple alignment with the current cluster members receives a high enough score (more than 30% similarity in our example). Once we have tested all the nodes of a given star, we proceed to the next star and in the same manner try to create a new cluster according to its edges. Figure 9(a) illustrates the process of creation of three clusters. The procedure terminates when all the nodes have been assigned to a cluster. Each cluster defines a consensus 3D pattern, which are the 3D points shared by all of its members. The coordinates that describe this pattern are taken from the node that has originated the cluster.
Prediction of dinucleotide binding sites
We have developed a method, RnaPred, which searches a complete protein for regions similar to the created 3D consensus patterns. The input protein and the patterns are represented by the set of their corresponding pseudocenter points (see Section 2). The RnaPred algorithm consists of the following 3 stages: (1) Generation of a set of candidate transformations; (2) Defining the 1:1 correspondence between the matched points and scoring; (3) Clustering the solutions of each 3D pattern as well as of different patterns mapped to the same protein region. Finally, we describe the selection of top ranking solutions and the evaluation of prediction success.
Superimposition
The generation of all candidate transformations is done with the Geometric Hashing method59 which consists of two stages, preprocessing and recognition. At the preprocessing, each triplet of pseudocenters from the complete molecule is considered as a local reference frame. Then, the coordinates of the other pseudocenters, calculated with respect to this coordinate system, are stored in a Geometric Hash Table. The key to the hash table consists of the point coordinates and physico-chemical property. In the recognition stage the same process is repeated for each 3D consensus pattern. For each pair of reference frames, one from a 3D pattern and one from the query protein, we count the number of matched points. For each pattern, we consider the reference frames with a significant number of matched points and construct a set of candidate transformations that can superimpose the given pattern upon the entire query protein. This procedure has several advantages for our goal. First, all the information of the complete molecule is stored only once for all the 3D patterns. Second, we avoid the processing of points that cannot be matched under any transformation. This is especially important for our application since the complete molecule is significantly larger than the 3D consensus patterns.
Correspondence and Scoring
The 1:1 correspondence and the scoring are implemented in the same manner as in the RnaBind method. We ignore solutions that are too small and insignificant and align less than 30% of a 3D consensus pattern (i.e. the score of the alignment is less than 30% of the score of the pattern aligned to itself).
Clustering
First, for each 3D pattern we cluster similar solutions that superimpose it on similar spatial locations. This is achieved by applying efficient RMSD clustering, described by Rarey et al60, with a default threshold of 3Å. Since our goal is to retain only a small set of top ranking solutions, we need to cluster the solutions that align different 3D consensus patterns to similar protein regions. We define two mappings of different 3D consensus patterns to be similar if their corresponding match lists (defined by the 1:1 correspondence) are based on at least 70% of identical pseudocenters of the complete query molecule. When similar alignments are detected, we retain only a single solution with the highest score.
Top ranking solutions and the evaluation of success
The list of top ranking solutions contains a set of different proteins regions that are recognized to resemble some of the constructed 3D patterns. The solutions are ranked in decreasing order of their score. A dinucleotide binding site is considered to be correctly predicted if one of the top ranking solutions fulfils at least one of the following requirements: (1) the RMSD distance between the dinucleotides of the real complex and those predicted by the alignment is ≤ 3 Å; (2) There are at least four (or 30%) physico-chemical properties (pseudocenters) involved in protein-dinucleotide interactions of the specific site that were correctly predicted. The percentage of successfully predicted dinucleotide binding sites is the success rate of the method.
Since the algorithm performs simultaneous alignment against all the constructed patterns, it is extremely fast. Its average running time for searching a complete protein surface for the presence of all the dinucleotide consensus binding patterns measured during all the leave-one-out tests (see Results) is 3 minutes on a standard PC (AMD Opteron 242, 1593MHz).
Reconstruction of protein-RNA complexes
Here, we extend the RnaPred algorithm described in the previous section to the recognition of the longest consecutive set of alignments of 3D patterns to neighboring protein regions. Using the binding modes of the dinucleotides bound to these patterns allows us to predict the structure of the protein interacting RNA fragment. Specifically, given all possible alignments of all the created 3D consensus patterns to the query protein, we construct a graph in the following manner. Each alignment solution is a graph node. It is represented by the superimposed pattern and the corresponding 3D transformation. We connect two nodes by an edge only if these represent alignments of 3D patterns to two neighboring regions so that their bound dinucleotides can be combined to form a continuous RNA fragment. Specifically, let (a1, a2) and (b1, b2) represent the RNA nucleotide pairs bound to two nodes, which represent two aligned 3D patterns. An edge between two nodes is created if the following requirements are satisfied after the application of the corresponding 3D transformations: (1) They have a pair of overlapping nucleotide rings: dist(a2, b1) ≤ ε1; (2) They have a pair of distant rings: dist(a1, b2) > ε1 (i.e. the 3D patterns are aligned to different protein regions); (3) The planes of the overlapping rings are parallel to each other: angle(a2, b1) ≤ ε2. Using ε1 = 2.0 and ε2 = 20°, we ensure that the dinucleotide pairs from two 3D patterns can be combined to a consecutive RNA fragment. Given the constructed graph, we search for the longest and highest scoring path in it. The score of a path is defined in the following way. The score of each node is the score of the alignment of the 3D physico-chemical pattern to a protein region. The score of a path is the sum of scores of its nodes. The length of a path is the number of nodes in it. One path is considered to be better than the other if it has at least the same length and its score is higher. The longest and the highest scoring path represents the alignment of the largest number of 3D patterns to neighboring protein regions that can allow the construction of an RNA fragment. Figure 9(b) provides a schematic representation of the main idea of this method.
Database of small molecule binding sites
The dataset of PDB structures complexed with small molecules was retrieved from the PDBsum database61, May 2007. We retained only the binding sites of compounds with more than 7 non-hydrogen atoms that are not covalently linked to the protein. In addition, to remove the binding sites complexed with natural substrates, such as ATP or GTP, we computed the frequency of occurrence of each ligand in the PDB. Small molecules that appeared in more than 10 complexes were assumed to be natural, frequently occurring substrates which can not make a significant contribution in our searches for drugs and drug fragments. As a result we constructed a dataset of 3999 binding sites which were screened for their similarity to nucleotide and dinucleotide binding patterns.
Supplementary Material
Supplementary Figure S1 presents histograms of angles and distances between the ring planes of consecutive, protein interacting nucleotides. Supplementary Table S1 presents the general frequencies of consecutive, extruded, protein interacting nucleotides that are not involved in RNA base pairing. Table S2 presents the analysis of the sequence specificity of pairs of consecutive, extruded π-stacked nucleotides. Table S3 presents the full list of clusters of single, extruded, nucleotide binding sites. Tables S4-S5 present the details of dinucleotide binding sites clusters of the AND-set and OR-set datasets respectively. Table S6 presents the details of the similarity between the Phe-Tyr binding platforms of HuD protein (1fxl) and Rho terminating factor (1pvo) revealed by cluster 23. Table S7 details the second type of the Phe-Tyr binding platform of RRMs and Rho termination factors (cluster 94). Table S8 describes the physico-chemical properties shared by dinucleotide binding sites of Pumilio protein (clusters 81–82). Table S9 details the physico-chemical properties shared by 3 consecutive dinucleotide binding sites of proteins with KH motifs (clusters 64, 41, 42).
Acknowledgments
We thank O. Dror for contribution of software for this project. The research of AS-P was supported by the Clore PhD Fellowship. The research of HJW has been supported in part by the Israel Science Foundation (grant no. 281/05), by the NIAID, NIH (grant No. 1UC1AI067231), by the Binational US-Israel Science Foundation (BSF)and by the Hermann Minkowski-Minerva Center for Geometry at TAU. This publication has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract NO1-CO-12400. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jones S, Daley DTA, Luscombe NM, Berman HM, Thornton JM. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadassy K, Wodak SJ, Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Kortemme T, Robertson T, Baker D, Varani G. A new hydrogen-bonding potential for the design of protein-RNA interactions predicts specific contacts and discriminates decoy. Nucleic Acids Res. 2004;32:5147–5162. doi: 10.1093/nar/gkh785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allers J, Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J Mol Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 5.Morozova N, Allers J, Myers J, Shamoo Y. Protein-RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics. 2006;22:2746–2752. doi: 10.1093/bioinformatics/btl470. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman MM, Khrapov MA, Cox JC, Yao J, Tong L, Ellington AD. AANT: the Amino Acid-Nucleotide Interaction Database. Nucleic Acids Res. 2004;32:D174–181. doi: 10.1093/nar/gkh128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treger M, Westhof E. Statistical analysis of atomic contacts at RNA-protein interfaces. J Mol Recognit. 2001;14:199–214. doi: 10.1002/jmr.534. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AC, Chen WW, Fuhrmann CN, Frankel AD. Recognition of nucleic acid bases and base-pairs by hydrogen bonding to amino acid side-chains. J Mol Biol. 2003;327:781–96. doi: 10.1016/s0022-2836(03)00091-3. [DOI] [PubMed] [Google Scholar]
- 9.Jeong E, Chung IF, Miyano S. A neural network method for identification of RNA-interacting residues in protein. Genome Inform. 2004;15:105–16. [PubMed] [Google Scholar]
- 10.Terribilini M, Lee JH, Yan C, Jernigan RL, Honavar V, Dobbs D. Prediction of RNA binding sites in proteins from amino acid sequence. RNA. 2006;12:1450–1462. doi: 10.1261/rna.2197306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, Dobbs D. RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007;35:W578–84. doi: 10.1093/nar/gkm294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim OTP, Yura K, Go N. Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 2006;34:6450–60. doi: 10.1093/nar/gkl819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006;34:W243–W248. doi: 10.1093/nar/gkl298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Draper DE. Themes in RNA-protein recognition. J Mol Biol. 1999;293:255–70. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- 15.Auweter SD, Oberstrass FC, Allain FHT. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klosterman PS, Hendrix DK, Tamura M, Holbrook SR, Brenner SE. Three-dimensional motifs from the SCOR, structural classification of RNA database: extruded strands, base triples, tetraloops and U-turns. Nucleic Acids Res. 2004;32:2342–52. doi: 10.1093/nar/gkh537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamura M, Hendrix DK, Klosterman PS, Schimmelman NR, Brenner SE, Holbrook SR. SCOR: Structural Classification of RNA, version 2.0. Nucleic Acids Res. 2004;32:D182–4. doi: 10.1093/nar/gkh080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Varani G. Protein families and RNA recognition. FEBS J. 2005;272:2088–97. doi: 10.1111/j.1742-4658.2005.04650.x. [DOI] [PubMed] [Google Scholar]
- 19.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murzin A, Brenner S, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 21.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–35. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sykes MT, Levitt M. Describing RNA structure by libraries of clustered nucleotide doublets. J Mol Biol. 2005;351:26–38. doi: 10.1016/j.jmb.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maris C, Dominguez C, Allain FHT. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS Journal. 2005;272 (9):2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 25.Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 26.Antson AA. Single-stranded-RNA binding proteins. Curr Opin Struct Biol. 2000;10:87–94. doi: 10.1016/s0959-440x(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 27.Turner B, Melcher SE, Wilson TJ, Norman DG, Lilley DM. Induced fit of RNA on binding the L7Ae protein to the kink-turn motif. RNA. 2005;11:1192–200. doi: 10.1261/rna.2680605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shajani Z, Drobny G, Varani G. Binding of U1A protein changes RNA dynamics as observed by 13C NMR relaxation studies. Biochemistry. 2007;46:5875–83. doi: 10.1021/bi602658x. [DOI] [PubMed] [Google Scholar]
- 29.Ellis JJ, Jones S. Evaluating conformational changes in protein structures binding RNA. Proteins. 2007 doi: 10.1002/prot.21647. in press. [DOI] [PubMed] [Google Scholar]
- 30.Moodie SL, Mitchell JBO, Thornton JM. Protein recognition of adenylate: an example of a fuzzy recognition template. J Mol Biol. 1996;263:486–500. doi: 10.1006/jmbi.1996.0591. [DOI] [PubMed] [Google Scholar]
- 31.Kuttner YY, Sobolev V, Raskind A, Edelman M. A consensus-binding structure for adenine at the atomic level permits searching for the ligand site in a wide spectrum of adenine-containing complexes. Proteins. 2003;52:400–411. doi: 10.1002/prot.10422. [DOI] [PubMed] [Google Scholar]
- 32.Connolly M. Analytical molecular surface calculation. J Appl Cryst. 1983;16:548–558. [Google Scholar]
- 33.Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence or fold homology. J Mol Biol. 2002;323:387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- 34.Rost B. Twilight zone of protein sequence alignments. Protein Engineering. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 35.Gardner PP, Wilm A, Washietl S. A benchmark of multiple sequence alignment programs upon structural rnas. Nucleic Acids Res. 2005;33:2433–2439. doi: 10.1093/nar/gki541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abraham M, Dror O, Nussinov R, Wolfson H. Classification and analysis of rna tertiary structures at three structural levels. 2008 doi: 10.1261/rna.853208. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–8. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, Bateman A. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiels JC, Tuite JB, Nolan SJ, Baranger AM. Investigation of a conserved stacking interaction in target site recognition by the U1A protein. Nucleic Acids Res. 2002;30:550–558. doi: 10.1093/nar/30.2.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grahn E, Moss T, Helgstrand C, Fridborg K, Sundaram M, Tars K, Lago H, Stonehouse NJ, Davis DR, Stockley PG, Liljas L. Structural basis of pyrimidine specificity in the MS2 RNA hairpin-coat-protein complex. RNA. 2001;7:1616–1627. [PMC free article] [PubMed] [Google Scholar]
- 41.Horn WT, Tars K, Grahn E, Helgstrand C, Baron AJ, Lago H, Adams CJ, Peabody DS, Phillips SE, Stonehouse NJ, Liljas L, Stockley PG. Structural basis of RNA binding discrimination between bacteriophages Qbeta and MS2. Structure. 2006;14:487–495. doi: 10.1016/j.str.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helgstrand C, Grahn E, Moss T, Stonehouse NJ, Tars K, Stockley PG, Liljas L. Investigating the structural basis of purine specificity in the structures of MS2 coat protein RNA translational operator hairpins. Nucl Acids Res. 2002;30 (12):2678–2685. doi: 10.1093/nar/gkf371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, McLachlan J, Zamore PD, Hall TM. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 44.Lewis HA, Musunuru K, Jensen KB, Edo C, Chen H, Darnell RB, Burley SK. Sequence-Specific RNA Binding by a Nova KH Domain: Implications for Paraneoplastic Disease and the Fragile X Syndrome. Cell. 2000;100:323–332. doi: 10.1016/s0092-8674(00)80668-6. [DOI] [PubMed] [Google Scholar]
- 45.Shulman-Peleg A, Nussinov R, Wolfson HJ. Recognition of functional sites in protein structures. J Mol Biol. 2004;339(3):607–633. doi: 10.1016/j.jmb.2004.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bunka DHJ, Stockley G. Aptamers come of age - at last. Nat Rev Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- 47.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 48.Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J. 2003;63 (3):751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higgins D, Thompson J, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer ELL. The Pfam Protein Families Database. Nucl Acids Res. 2002;30 (1):276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucl Acids Res. 2003;31 (17):5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 53.Shatsky M, Shulman-Peleg A, Nussinov R, Wolfson H. The multiple common point set problem and its application to molecule binding pattern detection. J Comput Biol. 2006;13:407–42. doi: 10.1089/cmb.2006.13.407. [DOI] [PubMed] [Google Scholar]
- 54.Shulman-Peleg A, Shatsky M, Nussinov R, Wolfson H. Spatial chemical conservation of hot spot interactions in protein-protein complexes. BMC Biology. 2007;5:43. doi: 10.1186/1741-7007-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denessiouk KA, Rantanen V, Johnson M. Adenine Recognition: A motif present in ATP-,CoA-,NAD-,NADP-, and FAD-dependent proteins. Proteins. 2001;44:282–291. doi: 10.1002/prot.1093. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr A. 1978;34:827–828. [Google Scholar]
- 57.Mehlhorn K. The LEDA platform of combinatorial and geometric computing. Cambridge: University Press; 1999. [Google Scholar]
- 58.Jain AK, Murty MN, Flynn PJ. Data clustering: a review. ACM Computing Surveys. 1999;3:264–323. [Google Scholar]
- 59.Wolfson HJ. Proc of the 1st European Conf on Comp Vision (ECCV) LNCS. Springer-Verlang; 1990. Model-Based Object Recognition by Geometric Hashing; pp. 526–536. [Google Scholar]
- 60.Rarey M, Wefing S, Lengauer T. Placement of medium-sized molecular fragments into active sites of proteins. J Computer-Aided Molecular Design. 1996;10:41–54. doi: 10.1007/BF00124464. [DOI] [PubMed] [Google Scholar]
- 61.Laskowski RA, Chistyakov VV, Thornton JM. PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 2005;33:D266–268. doi: 10.1093/nar/gki001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andreeva A, Howorth D, Murzin AG. The pre-SCOP database. 2007 June 4; http://www.mrc-lmb.cam.ac.uk/agm/pre-scop/
- 63.Lee TT, Agarwalla S, Stroud RM. A unique RNA Fold in the RumA-RNA-cofactor ternary complex contributes to substrate selectivity and enzymatic function. Cell. 2005;120:599–611. doi: 10.1016/j.cell.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 64.Agalarov SC, Sridhar-Prasad G, Funke PM, Stout CD, Williamson JR. Structure of the S15,S6,S18-rRNA complex: assembly of the 30S ribosome central domain. Science. 2000;288:107–13. doi: 10.1126/science.288.5463.107. [DOI] [PubMed] [Google Scholar]
- 65.Eiler S, Dock-Bregeon A, Moulinier L, Thierry JC, Moras D. Synthesis of aspartyl-tRNA(Asp) in Escherichia coli–a snapshot of the second step. EMBO J. 1999;18:6532–6541. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tishchenko A, Nikulin A, Fomenkova N, Nevskaya N, Nikonov O, Dumas P, Moine H, Ehresmann B, Ehresmann C, Piendl W, Lamzin V, Garber M, Nikonov S. Detailed analysis of RNA-protein interactions within the ribosomal protein S8-rRNA complex from the archaeon Methanococcus jannaschii. J Mol Biol. 2001;311:311–324. doi: 10.1006/jmbi.2001.4877. [DOI] [PubMed] [Google Scholar]
- 67.Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud R. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci USA. 2003;100:12648–53. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 presents histograms of angles and distances between the ring planes of consecutive, protein interacting nucleotides. Supplementary Table S1 presents the general frequencies of consecutive, extruded, protein interacting nucleotides that are not involved in RNA base pairing. Table S2 presents the analysis of the sequence specificity of pairs of consecutive, extruded π-stacked nucleotides. Table S3 presents the full list of clusters of single, extruded, nucleotide binding sites. Tables S4-S5 present the details of dinucleotide binding sites clusters of the AND-set and OR-set datasets respectively. Table S6 presents the details of the similarity between the Phe-Tyr binding platforms of HuD protein (1fxl) and Rho terminating factor (1pvo) revealed by cluster 23. Table S7 details the second type of the Phe-Tyr binding platform of RRMs and Rho termination factors (cluster 94). Table S8 describes the physico-chemical properties shared by dinucleotide binding sites of Pumilio protein (clusters 81–82). Table S9 details the physico-chemical properties shared by 3 consecutive dinucleotide binding sites of proteins with KH motifs (clusters 64, 41, 42).