

Abstract
Ribosome display was applied for affinity selection of antibody single-chain fragments (scFv) from a diverse library generated from mice immunized with a variant peptide of the transcription factor GCN4 dimerization domain. After three rounds of ribosome display, positive scFvs were isolated and characterized. Several different scFvs were selected, but those in the largest group were closely related to each other and differed in 0 to 5 amino acid residues with respect to their consensus sequence, the likely common progenitor. The best scFv had a dissociation constant of (4 ± 1) × 10−11 M, measured in solution. One amino acid residue in complementarity determining region L1 was found to be responsible for a 65-fold higher affinity than the likely progenitor. It appears that this high-affinity scFv was selected from the mutations occurring during ribosome display in vitro, and that this constitutes an affinity maturation inherent in this method. The in vitro-selected scFvs could be functionally expressed in the Escherichia coli periplasm with good yields or prepared by in vitro refolding. Thus, ribosome display can be a powerful methodology for in vitro library screening and simultaneous sequence evolution.
A number of evolutionary methods are currently being developed that can accelerate natural evolution of biological macromolecules to a matter of days. All of them have to fulfill two basic requirements: to couple genotype and phenotype for selection and to introduce diversification between selection rounds. Nucleic acids, where the molecules are simultaneously genotype and phenotype, have been evolved and selected for physical properties (1) or for binding to target molecules (2, 3). In contrast, most of the methods used for the selection of proteins as carrier of the phenotype have been based on living cells directly or indirectly by production of phages or viruses (4). However, in vivo approaches are limited by transformation efficiency (5), and the repeated construction of libraries with more than 109 to 1010 independent members is quite laborious. This limitation can be overcome by using in vitro systems based on cell-free translation.
Several in vitro selection approaches of polypeptides have been reported. Short peptides were affinity selected from a library by using polysomes (6, 7). Recently, using this concept, we developed a system, designated ribosome display, and we demonstrated that it is possible to carry out sequence evolution and phenotypic selection for ligand binding with a complete disulfide-containing protein: a single-chain fragment (scFv) of an antibody was enriched 108-fold in vitro by using ribosome display (8). Subsequently, it was reported that a scFv-κ construct of an antibody can also be selected by using a eukaryotic cell-free system (9) and that an in vitro synthesized polypeptide can be directly attached to its encoded message through a puromycin derivative that is synthetically coupled to the 3′ end of the mRNA (10, 11).
In the present study, we have prepared a murine antibody library, elicited against a monomeric variant of the yeast transcription factor GCN4. This protein is a member of the basic region leucine zipper family. It consists of an N-terminal activation domain, a basic DNA-binding domain, and a leucine zipper dimerization domain (12). Bound to its target site on the DNA, the dimeric GCN4 activates transcription of genes involved in amino acid biosynthesis (13).
We isolated and evolved GCN4-variant binding scFvs from this library by using ribosome display (8) and characterized them for affinity, folding, and expression in Escherichia coli. Most of the selected scFvs were closely related, and at least several appear to have been affinity matured during ribosome display. The best scFv had very high affinity to its antigen with a dissociation constant of around 10−11 M and a 65-fold improvement over its likely progenitor.
METHODS
Construction of Ribosome Display Library.
The variant peptide GCN4(7P14P), which contains two helix-breaking proline substitutions in surface exposed positions of the leucine zipper domain and possesses a random coil structure (data not shown), was prepared by chemical synthesis as described (14) and has the sequence x-RMKQLEPKVEELLPKNYHLENEVARLKKLVGER. For raising antibodies, three Balb/c mice were immunized, each with this antigen in a different format, administered in complete Freund’s adjuvant, namely (i) GCN4(7P14P) coupled to avidin through N-terminally linked biotin (x = biotinyl-GGG) (14), (ii) GCN4(7P14P) chemically crosslinked through its cysteine to lysines of keyhole limpet hemocyanine with N-succinimidyl 3-[2-pyridyldithio]-propionate (Pierce) (x = acetyl-CGGG), (iii) N-terminally disulfide-linked GCN4(7P14P) coupled to avidin through N-terminally linked biotin (x = biotinyl-GGGCGGG).
The three libraries, one from each mouse, were prepared separately, following the methods and primers described by Krebber et al. (15). In short, mRNA was extracted from about 1–5 × 106 spleen cells of immunized mice and transcribed to cDNA. After PCR amplification of the variable domains of the light chain (VL) and the variable domains of the heavy chain (VH), PCR products were purified by agarose gel electrophoresis and extracted from the gel with the QIAEX gel extraction kit (Qiagen). An assembly PCR was carried out (15) and the PCR products were directly diluted 3-fold in SfiI reaction buffer, digested with SfiI and separated by using agarose gel electrophoresis. The cut DNA was extracted from agarose gels by Amicon spin columns, concentrated by isopropyl alcohol precipitation and dissolved in sterile water. Purified PCR products (150 ng of each) were ligated in a 30-μl reaction mixture with SfiI-cut vector pAK200 (15) overnight at 16°C (molar ratio insert to vector = 1:2). To introduce the features necessary for ribosome display, the ligation mixtures were directly amplified in two steps by PCR, by using in the first step the primers SDA (5′-AGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAATATATCCATGGACTACAAAGA-3′), which introduced a ribosome binding site, and T3Te, which encodes the translated early transcription terminator of phage T3 (5′-GGCCCACCCGTGAAGGTGAGCCTCAGTAGCGACAG-3′), and in the second step primers T3Te and T7B (5′-ATACGAAATTAATACGACTCACTATAGGGAGACCACAACGG-3′), which introduced the T7 promoter as well as the 5′-loop (8). PCR products were used without purification for in vitro transcription, and RNA was purified by LiCl precipitation (16). The RNA from all three libraries was pooled in equal proportions and used for ribosome display.
In Vitro Translation of an scFv Antibody Library.
In vitro translations in an E. coli S-30 system were performed as described (8) with small modifications. In short, the in vitro translation was carried out for 8 min at 37°C in a 220-μl reaction that contained the following components: 50 mM Tris⋅HOAc, pH 7.5/30 mM NH4HOAc/12.3 mM Mg(OAc)2/0.35 mM of each amino acid/2 mM ATP/0.5 mM GTP/1 mM cAMP/0.5 mg/ml E. coli tRNA/20 μg/ml folinic acid/100 mM KOAc/30 mM acetylphosphate/1.5% polyethylene glycol 8,000/33 μg/ml rifampicin/1 mg/ml vanadyl ribonucleoside complexes/3.5 μM anti-ssrA oligonucleotide/0.3 μM protein disulfide isomerase/51.4 μl of E. coli MRE600 extract/90 μg/ml of mRNA.
Affinity Selection of Ribosome Complexes and RNA Isolation.
Affinity selection was performed as described previously (8) with small modifications. The translation was stopped by diluting it 4-fold with ice-cold washing buffer [50 mM Tris⋅HOAc pH 7.5/150 mM NaCl/50 mM Mg(OAc)2/2.5 mg/ml heparin/0.1% Tween 20] and centrifuged for 5 min at 4°C at 10,000 × g to remove insoluble components. Microtiter plates coated with BSA-GCN4(7P14P) conjugate were prewashed with ice-cold washing buffer, the supernatant from the centrifuged translation mixture, containing 2% sterilized milk, was applied (200 μl per microtiter well), and the plate was gently shaken for 1 hour at 4°C. After five washes with ice-cold washing buffer without heparin, the retained ribosome complexes were dissociated with ice-cold elution buffer (100 μl per well; 50 mM Tris⋅HOAc, pH 7.5/150 mM NaCl/10 mM EDTA/50 μg/ml S. cereviseae RNA) for 10 min at 4°C, and released mRNA was recovered by isolation by using the RNeasy kit (Qiagen). Purified RNA was used subsequently for reverse transcription–PCR (8). After in vitro transcription of PCR products, RNA was purified by LiCl precipitation (16) and used either for RIA analysis or for the next round of ribosome display.
RIA.
After each round of ribosome display, RNA of the whole pool was translated in vitro in an S-30 E. coli system by using similar conditions as described above for the library enrichment with the following modifications. The translation was carried out for 30 min at 37°C, the reaction mixture contained 0.3 μM of [35S]methionine (50 μCi/ml) and 0.35 mM each amino acid except methionine, and anti-ssrA oligonucleotide and protein disulfide isomerase were absent. After translation, the reaction mixture was diluted 4-fold with PBST (10 mM sodium phosphate buffer pH 7.4/140 mM NaCl/15 mM KCl/0.05% Tween 20) and centrifuged. The supernatant was diluted with the same volume of 4% milk in PBST containing 0 or 2 μM GCN4(7P14P) peptide and preincubated for 1 hour at room temperature. Binding to immobilized BSA-GCN4(7P14P) conjugate in microtiter wells was carried out for 30 min at 25°C with gentle shaking. After five washes with PBST, bound radioactive protein was eluted with 0.1 M triethylamine and quantified in a scintillation counter.
ELISA of Single scFvs.
After the third round of ribosome display, the PCR products were cloned into the vector pTFT74 (17). Plasmids of single clones were isolated and transcribed in vitro (16), RNA was purified by LiCl precipitation (16) and used for in vitro translation in an S-30 E. coli system by using similar conditions as described for the library above with the following modifications. Translation was carried out for 30 min at 37°C, without anti-ssrA oligonucleotide, vanadyl ribonucleoside complexes, and protein disulfide isomerase. After translation, the reaction mixture was diluted 4-fold with PBST and centrifuged.
The supernatant was diluted with the same volume of 4% milk in PBST containing 0, 2, 20, and 200 nM GCN4(7P14P) peptide and preincubated for 1 hour at room temperature. Binding to immobilized GCN4(7P14P) peptide in microtiter wells was carried out for 30 min at 25°C with gentle shaking, and bound scFv protein was detected by using the monoclonal anti-myc-tag antibody 9E10 and a polyclonal anti-mouse/peroxidase conjugate (Pierce). ELISA with purified proteins expressed in vivo (see below) was carried out analogously with 0.5, 5 and 50 ng scFv per well.
PCR Analysis of the Library for the Presence of the Asn-L34 → Ser Mutation.
The PCR products used for the preparation of the initial library (see above) were applied for PCR amplification, by using antisense primer VH3 annealing to both clones c11 and g5 (5′-GGCCCCAGTAGTCAAAGAGACCAG-3′) and a specific sense primer for the clone c11 C11VL1 (5′-CTGTTACAACTAGTAACTATGCCAA-3′) or a specific sense primer for the clone g5 G5VL1 (5′-CTGTTACAACTAGTAACTATGCCAG-3′). After amplification (4 min at 94°C, followed by 50 cycles of 30 sec at 94°C, 30 sec at 68°C and 2 min at 72°C finished by 10 min at 72°C), PCR products were analyzed by agarose gel electrophoresis.
Periplasmic Expression.
Selected scFv sequences were cloned in the secretion vector pAK400 (15). For expression, one liter of SB medium (20 g/l tryptone/10 g/l yeast extract/10 g/l NaCl/50 mM K2HPO4) containing 30 μg/ml chloramphenicol was inoculated with a preculture from a single bacterial colony and incubated at 25°C (5 liter flask, 160 rpm). Expression was induced at an A550 of 0.5 by addition of isopropyl D-thiogalactoside (Promega) to a final concentration of 1 mM. Incubation was continued for 5 hours. Cells were collected by centrifugation (8,000 × g, 10 min, 4°C) and resuspended in PBS, normalizing the cell densities to an A550 of 50. Cell disruption was achieved by French Press lysis, and the resulting crude extracts were centrifuged (20,000 × g, 15 min, 4°C). The supernatants were collected (soluble fraction) and pellets were resuspended in PBS containing 8 M urea (insoluble fraction).
Cytoplasmic Expression.
For cytoplasmic expression scFvs were cloned in the vector pTFT74 (17). For expression, one liter of SB medium containing 100 μg/ml ampicillin was inoculated with a preculture from a single bacterial colony of E. coli BL21(DE3) harboring the plasmid and incubated at 37°C (5 liter flask, 160 rpm). Expression was induced at an A550 of 1.0 by addition of isopropyl d-thiogalactoside to a final concentration of 1 mM. Incubation was continued for 4 hours. Cells were collected by centrifugation (8,000 × g, 10 min, 4°C), resuspended in 10 mM Tris⋅HCl, pH 8.0/2 mM MgCl2 and disrupted by sonication. Inclusion body protein was isolated following a standard protocol (18).
The inclusion body protein pellet from 1 liter of bacterial culture was solubilized at room temperature in 5 ml of solubilization buffer (0.2 M Tris⋅HCl, pH 8.0/6 M guanidinium hydrochloride/10 mM EDTA) containing 20 mM DTT. The resulting solution was centrifuged (48,000 × g, 10 min, 4°C) and about 2 ml of the supernatant (about 50 mg/ml of protein) was diluted in 1 liter of refolding buffer (0.2 M Tris⋅HCl/pH 9.0 at 4°C/0.8 M arginine/0.2 mM reduced glutathione/1 mM oxidized glutathione/2 mM EDTA), incubated for 2 days at 4°C and then applied to antigen affinity chromatography (see below). The overall yield of refolding and purification was about 10% of pure functional protein (for both scFvs) from total amount of protein used for refolding.
Antigen Affinity Chromatography.
To prepare the affinity matrix, biotinylated GCN4(7P14P) peptide was coupled to streptavidin agarose (Pierce). Crude extract from periplasmic expression, diluted 5-fold with 50 mM Tris⋅HCl, pH 8.0/500 mM NaCl, or refolding solution from inclusion bodies adjusted to pH 8.0 with HCl was directly loaded to the affinity column. After being washed with 50 mM Tris⋅HCl, pH 8.0/500 mM NaCl, bound scFv protein was eluted by using 0.1 M glycine⋅HCl, pH 2.2 and immediately neutralized with 2 M Tris. The eluates were concentrated by using Centricon columns (Amicon, cutoff 10,000 Da) and applied to a PD-10 column (Pharmacia) to exchange buffer to HBS (20 mM Hepes, pH 7.2/150 mM NaCl). The protein concentrations were calculated from the A280 values by using the calculated extinction coefficient (19).
Gel Permeation Chromatography.
Samples of purified and concentrated scFv proteins were analyzed on a Superose 12 column equilibrated with PBS on a SMART system (Pharmacia). The sample volumes were 50 μl containing 5 to 10 μg of protein, and the flow rate was 50 μl per min. Lysozyme (14 kDa), carbonic anhydrase (31 kDa), BSA (66 kDa), aldolase (150 kDa), and catalase (230 kDa) were used as molecular mass standards.
Western Blots.
Samples of soluble and insoluble fractions (see above) were separated by SDS/PAGE and blotted on poly(vinylidene difluoride) membranes. For immunodetection, bacterial crude extract containing an anti-His-tag scFv antibody fused to alkaline phosphatase (20) and the chemiluminescent substrate disodium 3-(4-methoxyspiro {1,2-dioxetane-3,2′-5′-chloro)tricyclo [3.3.1.13,7]decan}-4-yl)phenylphosphate (CSPD) (Boehringer Mannheim) were used.
Determination of the Antigen Dissociation Constant in Solution by Competition BIAcore.
Competition BIAcore analysis was performed under mass-transport limitation as described previously (21, 22) by using a sensor chip CM5 (Pharmacia) coated with 15,000 resonance units of BSA-GCN4(7P14P) conjugate or with only BSA as a control. Each binding–regeneration cycle was performed with a constant flow rate of 25 μl/min by using HBST (20 mM Hepes, pH 7.2/150 mM NaCl/0.005% Tween 20). Samples of 250 μl of antibody in HBST, containing various amounts of antigen, were injected by means of the sample loop of the system, followed by regeneration of the surface by injection of 20 μl of 6 M guanidinium chloride in HBST. Inhibition studies were carried out by coincubation of antibodies with GCN4(7P14P) peptide at a series of concentrations for at least 1 hour at 4°C before injection. The lowest concentration of scFv protein used for competition BIAcore analysis that gave satisfactory results was 10−9 M (maximal response ≈100 resonance units). Data were evaluated by using BIAevaluation software (Pharmacia) and kaleidagraph (Synergy Software, Reading, PA). Slopes of the association phase of linear sensograms were plotted against the corresponding total antigen concentrations, and the dissociation constant was calculated by using Eq. 1:
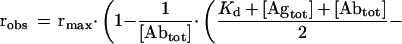 |
1 |
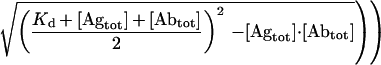 |
where robs is the slope at a given [Agtot], rmax is the maximal slope in the absence of inhibition by coincubated antigen, [Abtot] is the total antibody concentration, [Agtot] is the total antigen concentration, and Kd is the dissociation constant.
RESULTS AND DISCUSSION
Construction of the Library.
Mice were immunized with a variant of the GCN4 zipper, which contained two helix-breaking mutations (for sequence, see Methods) and thus was predominantly present as a random coil peptide (data not shown). Spleen mRNA was isolated, and VH and VL domains were amplified by reverse transcription–PCR and assembled in the orientation VL-(Gly4Ser)4-VH (15). This PCR amplification was the source of our initial diversity. To perform ribosome display, the scFv needed a tether at the C-terminal end to emerge from the ribosome tunnel so that folding could proceed without interference from the ribosome. We first ligated the library to the vector pAK200 (without transforming E. coli) to create a C-terminal fusion to the scFvs with part of gene III of filamentous phage M13 mp19 (15), which served as a tether. Ligation of the library to the vector pAK200 was very efficient: by agarose gel electrophoresis and restriction analysis, we found that all library scFv DNA was ligated at least to one side of the vector and more than 50% formed the required C-terminal fusion (data not shown).
By PCR of the ligation reaction we introduced, in the next step, stem loops that stabilize mRNA against exonucleases and sequences necessary for efficient transcription and translation. The final library construct contained a T7 promoter, 5′-stem loop, and Shine–Dalgarno sequence upstream of the scFv coding sequence, and downstream of the scFv gene a spacer consisting of 129 bases of gene III of filamentous phage M13 mp19 (amino acids 250–293), followed by 21 bases of translated T3Te, which was engineered to simultaneously constitute a 3′-stem loop (8).
Screening of the Library for GCN4(7P14P) Binders.
In the first approach (Fig. 1, experiment A), the affinity selection was performed as described for a model system (8) with the additional improvements of using heparin and sterilized milk during the affinity selection. We observed that these components decrease unspecific binding of ribosome complexes to antigen-containing as well as to control surfaces (data not shown). After the third round of ribosome display we analyzed the pool of scFvs by RIA for binding to the GCN4(7P14P) peptide. We found that the pool showed binding and nearly complete inhibition by 1 μM GCN4(7P14P) peptide (Fig. 1, experiment A). Analysis of single scFvs of this pool by ELISA revealed that approximately 75% of the scFvs were positive (data not shown).
Figure 1.

Analysis of pools after the third round of ribosome display. Pools of selected RNA were translated in vitro in the presence of [35S]methionine, and translation mixtures containing 0 and 1 μM GCN4(7P14P) peptide antigen as inhibitor were analyzed by RIA as described in Methods. Each bar represents the average of three samples. The ribosomal complexes were enriched on immobilized peptide antigen in the presence (experiment A) or in the absence (experiment B) of 2% sterilized milk during selection (for details, see text).
In the second approach (Fig. 1, experiment B), the same library was applied for ribosome display, but affinity selection was performed in the absence of milk. The obtained pool of scFvs after the third round of ribosome display gave about four times less bound protein than the pool obtained in experiment A and was nearly completely inhibited by 1 μM peptide (in the presence of milk in the RIA) (Fig. 1, experiment B). Analysis of single scFvs by ELISA revealed that of 24 scFvs only three bound the immobilized GCN4(7P14P) peptide, and binding could be inhibited with soluble peptide, while the other scFvs bound to the surface unspecifically (data not shown).
Analysis of Single scFvs.
Twenty-three binding scFvs from experiment A and three binders from experiment B were analyzed. Sequencing analysis revealed that of 26 scFvs, 22 were closely related and differed only in a few amino acid residues. The sequences of the other four scFvs were all different from each other and from the group of 22 related scFvs (data not shown), demonstrating functional diversity in the initial library. RIA analysis of these four scFvs showed lower binding to GCN4(7P14P) peptide, and inhibition with it was lower in comparison to the group of 22 related scFvs (see below).
Further analysis was thus performed with the group of 22 related scFvs. Sequence analysis showed that 20 of them were probably originating from one mRNA species, while two scFvs, designated c17 and c22, came from another mRNA (Fig. 2). ScFvs c17 and c22 contained six amino acid residues (11 bases) in complementarity determining region (CDR)2 and framework 3 of VH that were different from the other 20 scFvs (Fig. 2), too many to be introduced by PCR (24). The DNA sequence analysis of the 20 closely related scFvs showed that they contained between 4 and 13 base changes with respect to their consensus sequence. Some of these changes were introduced in framework 1 by the primer mixtures used for VH or VL PCR amplification. Excluding such changes, the scFvs contained 0–5 changed amino acid residues (2–8 bases on the DNA level) within the VH and VL domains. Zero to three changes occurred in the linker, where five scFvs contained a one-amino acid deletion (c2, c4, c7, c9, and g14) and one contained a 10-residue deletion (c1). Most mutations found in the linker were Gly to Ser and Gly to Asp substitutions (data not shown). We identified one scFv, designated c11, with no amino acid change with respect to the consensus sequence of the group, with the exception of the first amino acid residue of VH (Fig. 2), and we named this therefore the “consensus sequence” scFv c11.
Figure 2.

Alignment of amino acid sequences of related scFvs binding to GCN4(7P14P) peptide, isolated by ribosome display. Only VL and VH are shown. Residues identical to the consensus sequence of the 22 related scFvs (cons) are represented by dashes. The upper three scFvs (g2, g5, and g14) were isolated in experiment B, and the other 19 scFvs (c1 to c23) in experiment A (see Fig. 1). Numbering of amino acid residues in VL and VH and the labeling of CDRs is according to Kabat et al. (23).
Eleven scFvs, ten from the group of related sequences and one scFv that had no sequence similarity to the group of 22 related scFvs, were further analyzed by RIA for binding and inhibition by soluble peptide (Fig. 3). All selected scFvs bound to GCN4(7P14P) peptide and could be inhibited with 1 nM peptide to different levels. The best of them, scFv g5, showed the highest RIA binding signal as well as the highest inhibition level (Fig. 3). In contrast, the “consensus sequence” scFv c11 showed one of the lowest RIA binding signals as well as inhibition levels (Fig. 3). The four scFvs that had no sequence similarity to the group of 20 related scFvs all showed weaker binding and lower inhibition levels. The best of them, scFv c8, is shown for comparison in Fig. 3.
Figure 3.

RIA analysis of selected scFvs binding to GCN4(7P14P) peptide isolated by ribosome display and c11L34Ser mutant. RNA of single scFvs was translated in vitro in the presence of [35S]methionine, and translation mixtures containing 0, 1, 10, and 100 nM GCN4(7P14P) peptide were analyzed by RIA as described in Methods. Each bar represents the average of three samples. For experiments A and B, see Fig. 1.
Single-chain fragments g5 and c11 differed not only in several amino acid residues that were either introduced by oligonucleotides during library construction by PCR or were located in the linker, but also at position L34, which was changed from Asn (consensus) to Ser (selected). To prove our hypothesis of the importance of serine at position L34, we constructed the single Asn(L34)Ser substitution in the “consensus sequence” scFv c11 and designated it c11L34Ser. RIA analysis showed that, indeed, the c11L34Ser mutant behaved the same as scFv g5: it produced the same RIA signal as well as the same high level of inhibition with soluble peptide as scFv g5 (Fig. 3). These results confirmed that the single Asn(L34)Ser mutation was responsible for the better properties of scFv g5.
We produced c11 and c11L34Ser proteins in cytoplasmic inclusion bodies in E. coli, refolded them and purified the scFv proteins by using antigen affinity chromatography. Gel permeation chromatography revealed that both proteins were monomeric (data not shown). Equilibrium denaturation curves revealed stable proteins with a midpoint of denaturation of 1.9 M guanidinium chloride for the scFv c11 and 2.15 M guanidinium chloride for the c11L34Ser mutant, respectively. ELISA with these purified proteins (data not shown) showed a similar difference, as observed already by RIA of the proteins produced in vitro: mutant c11L34Ser gave 10-fold higher signals for binding than did the “consensus sequence” scFv c11, by using the same amount of antigen affinity-purified protein, and thus this difference in ELISA signal was very likely due to differences in the antigen affinity of these proteins.
Purified c11 and c11L34Ser proteins were used for Kd determination in solution by competition BIAcore analysis (21, 22). In this experiment, scFv protein was incubated with soluble antigen, and the mixture was injected on a BIAcore chip containing immobilized antigen. Only free scFv, but not antigen-bound scFv, could bind to antigen on the surface, analogous to the Friguet–Goldberg ELISA (25). The observed mass-transfer limited rates thus indicated the amount of free scFv in solution as a function of antigen concentration. Thereby, the correct dissociation constant in solution was obtained, independent of any BIAcore rebinding errors (26). The above-mentioned conditions resulted in a linear dependence of the observed rate on the amount of free scFv (data not shown). From a plot of the slopes against the corresponding total antigen concentration, Kd of the “consensus sequence” scFv c11 was calculated as (2.6 ± 0.1) × 10−9 M and that of the c11L34Ser mutant as (4 ± 1) × 10−11 M (Fig. 4).
Figure 4.

Determination of antigen dissociation constants (Kd) of c11 and c11L34Ser scFvs. Purified proteins c11 (2 nM) and c11L34Ser (1 nM) were mixed with different concentration of GCN4(7P14P) peptide and incubated for 1 hour before analysis. Samples were injected over the sensor chip coated with BSA-GCN4(7P14P) conjugate. From the linear sensograms, the slopes (resonance units vs. time in sec) were plotted against the corresponding total soluble antigen concentration. The slopes correlate to uncomplexed scFv in the injected solutions. From a fit with Eq. 1, Kd was calculated. Each point is the average of four independent experiments.
We also recloned both c11 and mutant c11L34Ser to the secretion vector pAK400 (15) and expressed both proteins in the periplasm of E. coli. The analysis of the expressed protein by Western blotting revealed that almost all of c11 and of c11L34Ser mutant was produced as soluble protein, and from one liter of culture about 1.5–2 mg of pure c11 or c11L34Ser protein was isolated by antigen affinity chromatography (data not shown). This result indicates that the proteins we have isolated by the in vitro method of ribosome display behave very well when functionally expressed in vivo in E. coli.
Evidence for Affinity Maturation in Vitro.
The light chain of the group of related scFvs is of mouse λ type and is unequivocally the product of the Vλ1 gene and the Jλ1 segment (27). The “consensus sequence” scFv c11 is identical to the unaltered Vλ1/Jλ1 gene sequence (27), and the AsnL34Ser mutation in scFv g5 might have been introduced already during affinity maturation in the mouse. We believe, however, that the AsnL34Ser mutation was introduced only during the ribosome display experiment. There are several facts that support our hypothesis. In both experiments A and B the same initial DNA pool was used. However, scFv g5 was isolated only in experiment B. If scFv g5 had been present in the initial pool, it should have been enriched also in experiment A, because the amount of antigen-bound protein is much higher for this scFv than for the other scFvs (Fig. 3). Additionally, scFv g5 was isolated only in the experiment where eight times fewer positive scFvs were found, suggesting it was only generated there. We also analyzed the initial library by PCR for the presence of the Asn to Ser mutation, using a specific primer that annealed to CDR1 of VL of scFv g5 DNA and that contained at the 3′ end a base matching only the mutated but not the consensus sequence. After 50 cycles of amplification, no PCR products were detected by agarose gel electrophoresis, while with another primer, which annealed to the same region of the consensus sequence, a very substantial PCR product of the expected size was found. All these facts together support the hypothesis that scFv g5 was generated during ribosome display in vitro. Interestingly, most of the selected scFvs (Fig. 3) performed better than the “consensus sequence” scFv c11. Therefore, ribosome display appears to be a very powerful method for the in vitro evolution of proteins.
Conclusions.
In this paper, we have shown that a diverse library of complex folded proteins can be screened for ligand binding entirely in vitro by using ribosome display, a method that does not use cells in any step. We showed that from a library of single-chain antibodies several proteins could be isolated that bound the antigen, the best of which with a Kd of 4 × 10−11 M. Because the protein does not have to be eluted from the ligand, as the RNA can be easily isolated from the bound ribosome complexes, the binding of the antibody to the target is not an upper limit to the affinity that can be selected. We have shown that the proteins we enriched by ribosome display can also be produced in vivo, either by refolding from inclusion bodies or by periplasmic expression, yielding reasonable amounts of protein that can be used for further analysis.
To generate large libraries, for example for phage display, is a very laborious and time-consuming process because of the many electroporations necessary (28). The advantage of ribosome display is that libraries containing 1012 or more independent functional members can be prepared easily and repeatedly after each randomization step and used for affinity selection. In ribosome display, proteins can also mutate during selection when DNA polymerases without proofreading function are used in PCR. Diversification of the proteins during selection can be increased even more when such methods as DNA shuffling (29), error-prone PCR (30), or the staggered extension process (31) are used. On the other hand, if mutation of proteins during the selection is not desired, proofreading polymerases can be used.
To include diversification steps with in vivo selection methods, such as phage display, requires either the use of mutator strains (32) or switching repeatedly between in vitro diversification and in vivo selection. The latter is laborious and not very convenient, and only a few studies (see refs. 33–35) have carried protein optimization through more than one generation. Taking together all advantages, namely screening of very large protein libraries for affinity to the ligand, performing the entire procedure in vitro without using any cells, and automatic diversification during the procedure, ribosome display can become a very powerful method for the directed evolution of proteins.
Acknowledgments
We thank Annemarie Honegger and Marcus Jäger for helpful discussions. This work was supported by Schweizerischer Nationalfonds Grants 3100-046624.96/1 and 3100-045556.95 and by a Kekulé fellowship from Fonds der Chemischen Industrie (Frankfurt, Germany) to L. J.
ABBREVIATIONS
- scFv
single-chain fragment of an antibody
- VL
variable domain of the light chain
- VH
variable domain of the heavy chain
- T3Te
early terminator of phage T3
- CDR
complementarity determining region
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Saffhill R, Schneider-Bernloehr H, Orgel L E, Spiegelman S. J Mol Biol. 1970;51:531–539. doi: 10.1016/0022-2836(70)90006-9. [DOI] [PubMed] [Google Scholar]
- 2.Gold L, Polisky B, Uhlenbeck O, Yarus M. Annu Rev Biochem. 1995;64:763–797. doi: 10.1146/annurev.bi.64.070195.003555. [DOI] [PubMed] [Google Scholar]
- 3.Irvine D, Tuerk C, Gold L. J Mol Biol. 1991;222:739–761. doi: 10.1016/0022-2836(91)90509-5. [DOI] [PubMed] [Google Scholar]
- 4.Phizicky E M, Fields S. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dower W J, Cwirla S E. In: Guide to Electroporation and Electrofusion. Chang D C, Chassy B M, Saunders J A, Sowers A E, editors. San Diego: Academic; 1992. pp. 291–301. [Google Scholar]
- 6.Mattheakis L C, Bhatt R R, Dower W J. Proc Natl Acad Sci USA. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gersuk G M, Corey M J, Corey E, Stray J E, Kawasaki G H, Vessella R L. Biochem Biophys Res Commun. 1997;232:578–582. doi: 10.1006/bbrc.1997.6331. [DOI] [PubMed] [Google Scholar]
- 8.Hanes J, Plückthun A. Proc Natl Acad Sci USA. 1997;91:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He M, Taussig M J. Nucleic Acids Res. 1997;25:5132–5134. doi: 10.1093/nar/25.24.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts R W, Szostak J W. Proc Natl Acad Sci USA. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemoto N, Miyamoto-Sato E, Husimi Y, Yanagawa H. FEBS Lett. 1997;414:405–408. doi: 10.1016/s0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- 12.Landschulz W H, Johnson P F, McKnight S L. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 13.Hope I A, Struhl K. Cell. 1985;43:177–188. doi: 10.1016/0092-8674(85)90022-4. [DOI] [PubMed] [Google Scholar]
- 14.Leder L, Berger C, Bornhauser S, Wendt H, Ackermann F, Jelesarov I, Bosshard H R. Biochemistry. 1995;34:16509–16518. doi: 10.1021/bi00050a035. [DOI] [PubMed] [Google Scholar]
- 15.Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard H R, Plückthun A. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 16.Pokrovskaya I D, Gurevich V V. Anal Biochem. 1994;220:420–423. doi: 10.1006/abio.1994.1360. [DOI] [PubMed] [Google Scholar]
- 17.Ge L, Knappik A, Pack P, Freund C, Plückthun A. In: Antibody Engineering. Borrebaeck C A K, editor. New York: Oxford Univ. Press; 1995. pp. 229–266. [Google Scholar]
- 18.Buchner J, Rudolph R. Biotechnology. 1991;9:157–164. doi: 10.1038/nbt0291-157. [DOI] [PubMed] [Google Scholar]
- 19.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 20.Lindner P, Bauer K, Krebber A, Nieba L, Kremmer E, Krebber C, Honneger A, Klinger B, Mocikat R, Plückthun A. BioTechniques. 1997;22:140–149. doi: 10.2144/97221rr01. [DOI] [PubMed] [Google Scholar]
- 21.Karlsson R. Anal Biochem. 1994;221:142–151. doi: 10.1006/abio.1994.1390. [DOI] [PubMed] [Google Scholar]
- 22.Nieba L, Krebber A, Plückthun A. Anal Biochem. 1996;234:155–165. doi: 10.1006/abio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 23.Kabat E A, Wu T T, Perry H M, Gottesmann K S, Foeller C. Sequences of Proteins of Immunological Interest. I. Bethesda, MD: U.S. Department of Health and Human Services; 1991. p. 151. and 464, 5th Ed. [Google Scholar]
- 24.Keohavong P, Thilly W G. Proc Natl Acad Sci USA. 1989;86:9253–9257. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friguet B, Chaffotte A F, Djavadi-Ohaniance L, Goldberg M E. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 26.Schuck P. Annu Rev Biophys Biomol Struct. 1997;26:541–566. doi: 10.1146/annurev.biophys.26.1.541. [DOI] [PubMed] [Google Scholar]
- 27.Eisen H N, Reilly E B. Annu Rev Immunol. 1985;3:337–365. doi: 10.1146/annurev.iy.03.040185.002005. [DOI] [PubMed] [Google Scholar]
- 28.Vaughan T J, Williams A J, Pritchard K, Osbourn J K, Pope A R, Earnshow J C, McCafferty J, Hodits R A, Wilton J, Johnson K S. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 29.Stemmer W P. Nature (London) 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 30.Cadwell R C, Joyce G F. PCR Methods Appl. 1994;3:S136–S140. doi: 10.1101/gr.3.6.s136. [DOI] [PubMed] [Google Scholar]
- 31.Zhao H, Giver L, Shao Z, Affholter J A, Arnold F H. Nat Biotechnol. 1998;16:258–261. doi: 10.1038/nbt0398-258. [DOI] [PubMed] [Google Scholar]
- 32.Low N M, Holliger P, Winter G. J Mol Biol. 1996;260:359–368. doi: 10.1006/jmbi.1996.0406. [DOI] [PubMed] [Google Scholar]
- 33.Yang W P, Green K, Pinz-Sweeney S, Briones A T, Burton D R, Barbas C F., 3rd J Mol Biol. 1995;254:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
- 34.Schier R, McCall A, Adams G P, Marshall K W, Merritt H, Yim M, Crawford R S, Weiner L M, Marks C, Marks J D. J Mol Biol. 1996;263:551–567. doi: 10.1006/jmbi.1996.0598. [DOI] [PubMed] [Google Scholar]
- 35.Braisted A C, Wells J A. Proc Natl Acad Sci USA. 1996;93:5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]