

Abstract
Congenital generalized lipodystrophy, also known as Berardinelli-Seip syndrome, is a very rare hereditary syndrome that is characterized by an almost complete absence of adipose tissue from birth. Cardiac involvement seems to have substantial influence in the long-term prognosis.
Herein, we report an apparently unique case of congenital generalized lipodystrophy with cardiac sequelae. A 17-year-old woman, diagnosed in childhood with Berardinelli-Seip syndrome, presented with severe epigastric pain that was secondary to previous myocardial infarction. The patient had ischemia, dilated cardiomyopathy, and congestive heart failure, but no coronary artery disease. She was discharged from the hospital in stable condition after 3 days of medical treatment.
To our knowledge, this is the 1st reported case of congenital generalized lipodystrophy with dilated cardiomyopathy, congestive heart failure, severe mitral regurgitation, and inferior myocardial infarction as cardiac sequelae of this syndrome—but without evidence of coronary artery disease or cardiac hypertrophy. In addition to discussing this patient's case, we present diagnostic and therapeutic approaches to Berardinelli-Seip syndrome.
Key words: Cardiomyopathy, hypertrophic/diagnosis/etiology/physiopathology; diabetes mellitus, lipoatrophic/complications; genetic markers; heart diseases/diagnosis; hyperlipidemias/genetics; lipodystrophy/congenital/complications/epidemiology/genetics/therapy
Coronary artery disease is not one of the usual cardiac sequelae of congenital generalized lipodystrophy (CGL), also known as Berardinelli-Seip syndrome. Herein, we report the case of a young woman who presented with epigastric pain secondary to inferior myocardial infarction from CGL. Although the patient had CGL with cardiac sequelae, she had no coronary artery disease and no cardiac hypertrophy. We discuss the apparently unique case of this patient and various diagnostic and therapeutic approaches to CGL.
Case Report
In June 2001, a 17-year-old black American woman with a history of CGL and recurrent epigastric pain was admitted to the hospital because of sharp, localized epigastric pain. She had no fever, chest pain, shortness of breath, cough, or other gastrointestinal symptoms. Her medical history included type I diabetes mellitus with 4 years of severe insulin resistance, recurrent acute pancreatitis, mesenteric syndrome, and mixed hyperlipidemia. She had no family history of lipodystrophy, and no personal or family history of heart disease. She had been diagnosed with CGL in childhood, on the basis of acromegaloid features, the absence of adipose tissue on physical examination, a fatty liver and hepatomegaly on abdominal ultrasonography, diabetes mellitus, hyperlipidemia, and a skeletal survey of long bones that showed osteopenia with bone cysts. Her medications included subcutaneous isophane insulin (3 UI/[kg·d]), and oral metformin (1,000 mg twice daily), gemfibrozil (1,200 mg twice daily), and simvastatin (60 mg nightly).
Physical examination revealed a thin young woman with an acromegaloid appearance and a lack of subcutaneous fat. She also lacked buccal fat and had no jugular venous distention. Her vital signs were normal. Her heart was in regular rhythm with a 3/6 systolic murmur, mostly apical. Auscultation of the lungs revealed bilateral basal crackles. Her abdomen was soft, with a prominent umbilicus, epigastric tenderness, and mild hepatomegaly. She had thin limbs with trace leg edema, hirsutism, and acanthosis nigricans in the groin and axilla. She had a normal IQ, and her neurologic examination was normal. She also had enlarged genitalia and irregular menstrual cycles.
The laboratory findings were unremarkable, with normal levels of cardiac enzymes. Abdominal ultrasonography showed a fatty liver with homogeneous hepatomegaly. Chest radiography revealed cardiomegaly with bilateral pulmonary congestion and mild bibasilar pleural effusion. Electrocardiography showed sinus rhythm, left-axis deviation, poor R-wave progression, small Q waves with T-wave inversion, and slight ST-segment elevation in leads V3 through V6. Echocardiography showed a left ventricular ejection fraction (LVEF) of 0.24, an enlarged left atrium (volume, 65 mL) and LV (LV end-diastolic diameter, 5.9 cm), normal LV wall thickening, severe inferolateral and mild anterior LV wall hypokinesis, and severe mitral insufficiency. A thallium stress test showed an inferolateral fixed defect that was consistent with previous myocardial infarction.
Diagnostic cardiac catheterization was performed. The pertinent findings were mild-to-moderate depression of left ventricular systolic function, with an LVEF of 0.40 to 0.50; and moderate-to-severe hypokinesis of the inferior segment, with mild hypokinesis of the anterior and apical segments. Only the anterobasilar segment contracted normally. Mitral regurgitation was severe, causing the LVEF to appear to be better than it was. Her coronary arteries were normal. No other imaging techniques were applied. Pulmonary capillary wedge tracing showed a large V wave with elevation of pulmonary artery pressure to 50/25 mmHg. The pulmonary artery wedge pressure was 31 mmHg.
We determined that the epigastric pain was secondary to mesenteric syndrome or, alternatively, was stable angina due to ischemia from the old inferior myocardial infarction. The patient also had congestive heart failure with dilated cardiomyopathy. Pancreatitis and acute myocardial infarction were ruled out because of the normal levels of cardiac enzymes, amylase, and lipase. After 3 days of treatment with angiotensin-converting enzyme inhibitors, diuretics, and aspirin, the patient was discharged from the hospital in stable condition. At a follow-up examination 3 months after discharge from the hospital, she remained in stable condition on medical therapy.
Discussion
History, Prevalence, and Origin of Berardinelli-Seip Syndrome
Berardinelli-Seip syndrome (CGL) is an autosomal recessive disorder in which there is an almost complete absence of adipose tissue from birth. It was discovered by Berardinelli in 19541 and confirmed by Seip in 1959.2 Men and women are equally affected, and the clinical manifestations are usually obvious from birth. It is a very rare disorder with a prevalence of 1 per 10 million persons worldwide, 1 per 12 million in the United States, 1 per 1 million in Norway, 1 per 500,000 in Portugal, and 1 per 200,000 in Lebanon.3
The molecular mechanisms of Berardinelli-Seip syndrome are not fully understood. At least 3 loci have been implicated in different types of the disorder: CGL1 is due to mutations on the long arm of chromosome 9 (9q34), which cause changes in the enzyme AGPAT2 (1-acylglycerol-3-phosphate O-acyltransferase 2); CGL2 is a recently discovered, more severe type of CGL due to mutations in the gene encoding for seipin (BSCL2, Berardinelli-Seip congenital lipodystrophy 2), which is located on band 11q134; and CGL3, for which data are scarce and unclear, not linked to either 11q13 or 9q34.
Diagnosis and Epidemiology
The 1st step in the diagnostic procedure is to ascertain the family medical history of the patient (ideally, for at least 3 generations), the ethnicity of the patient's grandparents, and parental consanguinity. Initial diagnostic evaluation should include clinical features and physical findings, blood chemistry, abdominal ultrasonography, and echocardiography. More specific tests can also be performed, including a skeletal survey (especially of the long bones) to check for osteopenia and cysts and to check bone maturation. A complete ophthalmologic examination, Wechsler testing for IQ, and DNA testing are very helpful in confirming the diagnosis. Prenatal diagnosis can be performed by means of chorionic villi sampling between 9 and 12 weeks of gestation. Findings associated with CGL can be divided into major and minor diagnostic criteria3 (Table I).
TABLE I. Diagnostic Criteria of Berardinelli-Seip Syndrome

In all reported cases, hepatic dysfunction, diabetes mellitus, hypertrophic cardiomyopathy, and hyperlipidemia have been substantial contributors to morbidity and death.
Despite hyperlipidemia, atherosclerotic vascular complications are rare in patients who have CGL—to our knowledge, only 33 instances of cardiac disease have been reported in the medical literature.5–10 Van Maldergem and colleagues10 studied 70 CGL-affected individuals from 44 unrelated families. All of the studied patients had hypertrophy of the skeletal muscles. Results of cardiac ultrasonography were reported in some cases: 15 patients were diagnosed with hypertrophic cardiomyopathy (HCM), and only 2 patients had normal echocardiographic results. Heart failure was reported in 3 individuals as a cause of death.10
Typical cardiac findings are summarized in Table II. The cardiac manifestations in our patient were atypical for CGL: low LVEF, evidence of inferior myocardial infarction with normal coronary vessels, and no evidence of cardiac hypertrophy.
TABLE II. Cardiac Involvement in 33 Reported Cases5–10 of Berardinelli-Seip Syndrome
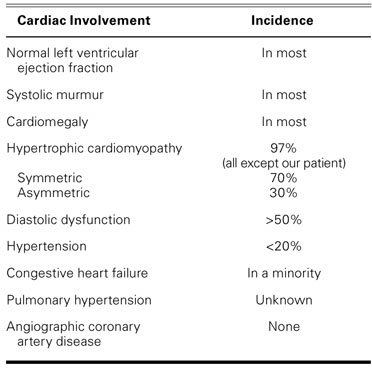
In autopsy studies, some patients with CGL were found to have had anteroseptal myocardial infarctions. In some histologic studies, nonspecific fibrotic changes and coronary artery disease were found in patients who had the syndrome. Comorbid HCM might be related to the action of insulin in the cardiac receptors of insulin-like growth factor (IGF-1).5 This comorbid HCM rarely leads to heart failure and differs histologically from classic HCM by the absence of fiber rearrangement.11
Treatment and Prognosis
Patients with CGL should avoid excessive food intake, undergo leptin-replacement therapy, and consider improving their physical appearance by means of cosmetic surgery. The treatment for dyslipidemia should involve an extremely low-fat diet (<15% of calories from fat), fibrates, fish oil, exercise, and no alcohol consumption. Hyperglycemia can be treated with metformin,12,13 highly concentrated insulin (500 U/mL) for patients who require more than 400 units of insulin per day, and insulin lispro.13 Recombinant IGF-1 has been shown to provide some benefit. Echocardiography and ultrasonography of the liver should be repeated every 6 months. Death occurs mostly in the 3rd decade of life from cirrhosis and its complications, recurrent acute pancreatitis, diabetic complications, and cardiac diseases.14
To our knowledge, our patient is the first to have dilated cardiomyopathy, congestive heart failure, severe mitral regurgitation, and inferior myocardial infarction without evidence of coronary artery disease as cardiac sequelae of Berardinelli-Seip syndrome. She had no evidence of cardiac hypertrophy on echocardiography or during catheterization, which is unusual for this disease.
In all previous reports, patients with generalized lipodystrophy syndrome (whether congenital or acquired) have had HCM as the chief cardiac sequela. Cardiac involvement seems to have an important influence on long-term prognosis. Further understanding of the molecular mechanisms that underlie CGL may lead to the discovery of therapeutic approaches that will prevent the loss of adipocytes, induce adipogenesis in lipodystrophic regions, and prevent or delay the onset of metabolic complications in patients with CGL.
Acknowledgments
We thank Dr. Monica Goble and Dr. Joel Eisenberg of the University of Michigan for their contributions to this paper, and the Scientific Publications department of the Texas Heart Institute for editorial assistance in the preparation of this manuscript.
Footnotes
Address for reprints: Wissam I. Khalife, MD, 4360 Staghorn Lane, Friendswood, TX 77546. E-mail: wkhalife@yahoo.com
References
- 1.Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab 1954;14(2): 193–204. [DOI] [PubMed]
- 2.Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr 1959;48:555–74. [PubMed]
- 3.Van Maldergem L. Berardinelli-Seip congenital lipodystrophy. Orphanet encyclopedia November 2001; 1–6. Available from: http://www.orpha.net/data/patho/GB/uk-berard.pdf
- 4.Magre J, Delepine M, Khallouf E, Gedde-Dahl T Jr, Van Maldergem L, Sobel E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 2001;28(4): 365–70. [DOI] [PubMed]
- 5.Geffner ME, Golde DW. Selective insulin action on skin, ovary, and heart in insulin-resistant states. Diabetes Care 1988;11(6):500–5. [DOI] [PubMed]
- 6.Klar A, Brand A, Hurvitz H, Gross-Kieselstein E, Branski D. Cardiomyopathy in lipodystrophy and the specificity spillover hypothesis. Isr J Med Sci 1993;29(1):50–2. [PubMed]
- 7.Bjornstad PG, Foerster A, Ihlen H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl 1996;413:39–43. [DOI] [PubMed]
- 8.Rheuban KS, Blizzard RM, Parker MA, Carter T, Wilson T, Gutgesell HP. Hypertrophic cardiomyopathy in total lipodystrophy. J Pediatr 1986;109(2):301–2. [DOI] [PubMed]
- 9.Viegas RF, Diniz RV, Viegas TM, Lira EB, Almeida DR. Cardiac involvement in total generalized lipodystrophy (Berardinelli-Seip syndrome) [in English, Portuguese]. Arq Bras Cardiol 2000;75(3):243–8. [DOI] [PubMed]
- 10.Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T Jr, Delepine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy [published erratum appears in J Med Genet 2003;40(2):150]. J Med Genet 2002;39(10):722–33. [DOI] [PMC free article] [PubMed]
- 11.Geffner ME, Santulli TV Jr, Kaplan SA. Hypertrophic cardiomyopathy in total lipodystrophy: insulin action in the face of insulin resistance? J Pediatr 1987;110(1):161. [DOI] [PubMed]
- 12.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet 2001;358(9285):893–4. [DOI] [PubMed]
- 13.Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med 1998; 338(26):1876–80. [DOI] [PubMed]
- 14.Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004;350(12):1220–34. [DOI] [PubMed]