

Nitrogen-containing heterocyclic systems have attracted considerable interest over the years because they form the core structures, and are key intermediates, of natural products.[1] One of the most appealing synthetic approaches for their preparation is the intramolecular hydroamination of alkenes, in which the nitrogen–carbon bond is formed by addition of an amine to an olefin.[2] Various catalysts have been used to effect this transformation, which include alkali metals,[3] early[4] and late transition metals,[5] and f-block elements.[6] Interestingly, despite the buffering effect of amines, intramolecular[7] and even intermolecular[8] acid-catalyzed hydroaminations have recently been developed. Schlummer and Hartwig[7a] reported the cyclization of amino alkenes bearing an electron-withdrawing group on the nitrogen atom by catalysis with triflic or sulfuric acid (20 mol %; Scheme 1). A mechanistic study of this process suggested that in contrast to similar transformations using various electrophiles as promoters (for example, iodine-,[9] bromine-,[10] and selenium-based electrophiles[11]), the first step was protonation of the amine, followed by intramolecular transfer of the proton to the double bond in the rate-determining step, and lastly trapping of the generated cation by the amino group. Accordingly, in the absence of electron-withdrawing groups on the nitrogen atom, the cyclization does not occur because of the excessive basicity of the amino group, which prevents the transfer of the proton to the olefin.
Scheme 1.

a) Schematic representation of acid-catalyzed intramolecular hydroamination; EWC = electron-withdrawing group, n = l,2. b) The hydroiminiumation reaction as a potential synthetic route to cyclic iminium salts A. c) CAAC/H+ salts A′, the precursors of CAACs B; R1, R2 ≠ H, Ar = bulky aryl group.
Imines are certainly less basic than amines, and therefore it was decided to investigate the feasibility of “hydroiminiumation” reactions, which would be an atom-economical route to cyclic iminum salts A (Scheme 1). Providing there is a bulky aryl substituent on the nitrogen atom and that there is a quaternary carbon atom in the position α to the aldiminium carbon atom, salts A′ are the direct precursors of stable cyclic alkyl amino carbenes (CAACs) B.[12] We have shown that CAACs can compete with N-heterocyclic carbenes (NHCs)[13] as ligands for transition-metal-based catalysts,[12a] and also allow the preparation of very low coordinate transition-metal centers.[12b] We report herein our preliminary results on the scope of the thermally induced hydroiminiumation reaction and its application to the synthesis of a variety of CAAC precursors.
To establish the viability of this hydroiminiumation methodology, the synthesis of the previously reported CAAC/H+ compound 4a[12a] was chosen as an initial test. Deprotonation of aldimine 1a, derived from 2,6-diisopropyl-aniline (DippNH2) and cyclohexane carboxaldehyde, with lithium diisopropylamide (LDA) leads to the corresponding 1-aza-allyl anion, which readily reacts at room temperature with 3-bromo-2-methylpropene (or 3-chloro-2-methylpropene) to afford the alkenyl aldimine 2a in 94% yield (Scheme 2). Addition of a stoichiometric amount of a 2M solution of HCl/Et2O to a toluene solution of 2a at −78 °C resulted in the immediate formation of a white precipitate. After 15 minutes at −78 °C, the mixture was allowed to warm to room temperature and stirring was continued for an additional 15 minutes. After filtration and recrystallization from chloroform, a new compound 3a was isolated as white crystals in 92% yield. The ionic character of 3a was apparent from its low solubility in toluene, while its acyclic nature was revealed by the presence of a 13C NMR signal at δ = 117.0 ppm from an ethylenic CH2 fragment. The protonation of the nitrogen atom was indicated by a 1H NMR signal at δ = 15.5 ppm, and by the deshielding of the N=CH 13C and 1H NMR signals (2a: (δ = 173.6 and 7.6 ppm; 3a: (δ = 189.8 and 8.0 ppm). A single-crystal X-ray diffraction study unambiguously proved the alkenyl aldiminium structure of 3a (Figure 1). [14] Pleasingly, it was noted that heating an aceto-nitrile solution of 3a in a tube sealed by a teflon stopcock at 50 °C for 18 h afforded the desired cyclic iminium salt 4a in 88% yield. Obviously, the last two steps of the synthesis (2a→4a) can be performed in situ, and the best results (88% yield of isolated product) were obtained when a twofold excess of HCl was used. The overall transformation (1a→4a) can thus be done in 83% yield, which compares extremely favorably with the previously reported method (48 % yield); moreover, the new route uses the same precursor 1a, but avoids the use of the costly reagents 1,2-epoxy-2-methylpropane and trifluoromethane sulfonic anhydride.
Scheme 2.

Influence of the nature of the R and R1 substituents on the rate of the hydroiminiumation reaction; Dipp = 2,6-iPr2C6H3; Mes = 2,4,6-Me3C6H2. [a] Time and temperature required for complete conversion of 3. [b] Yield of isolated product, without isolation of 3, and using a twofold excess of HCl. [c] n.r. = no reaction.
Figure 1.

Molecular structure of 3a in the solid state.
To study the influence of various steric and electronic factors on the hydroiminiumation reaction, several different alkenyl aldimines 2b–h were prepared (Schemes 2, 3, and 4). Without exception, the protonation occurred smoothly at the nitrogen atom, and the ensuing alkenyl aldiminium salts 3b–h were obtained in good to excellent yields. The cyclization process occurs slightly more easily when bulky substituents are used on both sides of the NCC fragment (Scheme 2). Indeed, when two methyl groups were used in place of the cyclohexyl group of 3a, the formation of 4b required 24 h at 50 °C, whereas for derivative 3c (Ar = Mes, CR1R1 = CMe2) 24 h at 70 °C are necessary to achieve complete conversion. Not surprisingly, a limitation to the methodology was found when an electron-donating tert-butyl group was placed on the nitrogen atom. Here, because of the high basicity of the nitrogen center, no trace of the cyclic iminium salt 4d was detected when a toluene solution of 3d was heated at 110°C for 24 h.
Scheme 3.

Influence of the nature of the alkene substituents R1 and R2 on the rate and regioselectivity of the hydroiminiumation reaction. Tos=toluene-4-sulfonyl. [a] Time and temperature required for complete conversion of 3. [b] Yield of isolated product, without isolation of 3, and using a twofold excess of HCl. [c] The reaction did not go to completion and was stopped after 72 h. [d] Yield as measured by NMR spectroscopy.
Scheme 4.
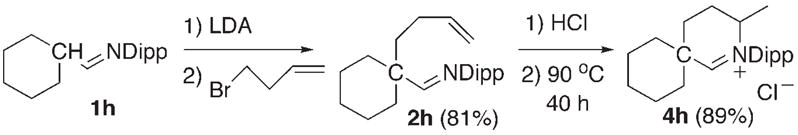
Synthesis of six-membered heterocyclic aldiminium salt 4h.
Use of alkenyl aldiminium salts 3a and 3e–g allowed study of the influence of the substitution pattern of the carbon–carbon double bond on the fate of the hydroiminiumation reaction, especially with regard to its regioselectivity (Scheme 3). The temperature required for cyclization was found to increase along the series 3a<3e<3f<3g. More importantly, in all cases five-membered heterocycles 4 resulting from exo cyclization were obtained, with no trace of the six-membered-ring isomers being detected. Strikingly, the cyclization of 3g affords exclusively five-membered heterocycle 4g (Figure 2), despite the presence of a phenyl group at the terminal carbon atom of the olefin, which would be expected to stabilize a benzylic carbocation intermediate. Together these observations favor a mechanism in which the proton would be transferred intramolecularly to the double bond in the rate-determining step, similarly to the mechanism proposed by Schlummer and Hartwig for the acid-catalyzed hydroamination reaction.[7a] When compared to the latter reaction involving alkenyl amine I, for which the formation of the six-membered ring II was observed, our result suggests that the addition of N—H across the double bond has a greater “concerted” character[15] in the hydroiminiumation than in the hydroamination reaction.[7a]
Figure 2.

Molecular structures of 3g (left) and 4g (right) in the solid state.
Six-membered heterocyclic aldiminium salts can also be accessed as shown by the preparation of 4h (Scheme 4). However, as observed in the hydroamination reaction,[7a] the cyclization to 4h is more difficult than for the homologous five-membered ring 4e.
Besides the easy preparation of a wide variety of CAAC/H+ compounds, the intramolecular hydroiminiumation reported here features some distinct advantages when compared to the intramolecular hydroamination reaction. The resulting iminium ions are very reactive, potentially allowing for the subsequent addition of a large range of nucleophiles, and since they are often prochiral, this chemistry offers the possibility of facile construction of a new stereogenic center a to the nitrogen atom. The extension of this work to other protonated sp2-nitrogen-containing species is under active investigation.
Experimental Section
All manipulations were performed under argon by using standard Schlenk techniques and oven-dried, argon-flushed glassware. Dry, oxygen-free solvents were employed. 1H and 13C NMR spectra were recorded on Varian Inova 300 and Bruker Avance 300 spectrometers.
Representative procedure for the synthesis of alkenyl imines 2: A solution of LDA (1.18 g, 11.0 mmol) in Et2O (20 mL), cooled to −78°C, was added to a solution of aldimine la (3.00 g, 11.0 mmol) in Et2O (20 mL) at −78 °C. After 15 minutes the mixture was left to warm to room temperature and stirring was continued for an additional two hours. The volatiles were then removed under vacuum to afford an oily yellow-orange residue, which was dissolved in Et2O (30 mL) and cooled to −78 °C. 3-Bromo-2-methylpropene (1.11 mL, 11.1 mmol) was then slowly added. After 15 minutes the solution was warmed to room temperature and stirring was continued for an additional 12 h. Removal of the volatiles under vacuum and extraction with hexanes afforded alkenyl aldimine 2a as a light-yellow oil in 94% yield. 13C{1H} NMR (75.1 MHz, CDCl3, 25°C): δ = 173.6 (N=CH), 149.1 (Cipso), 142.3 (C=CH2), 137.7 (Cortho), 123.9 (Cpara), 123.0 (Cmeta), 115.5 (=CH2), 46.6 (CCy), 44.5 (CH2), 33.5 (H2CCy), 27.7 (CHCH3), 26.1 (H2CCy), 25.6 (CH3C=), 23.8 and 23.6 (CHCH3), 22.8 ppm (H2CCy); 1H NMR (300.0 MHz, CDCl3, 25 °C): δ = 7.60 (s, 1H, CH=N), 7.18–7.09 (m, 3H, Haro), 4.95 (s, 1H, C=CH2), 4.80 (s, 1H, C=CH2), 3.06 (sept, JHH = 6.8Hz, 2H, CHCH3), 2.35 (s, 2H, CH2), 1.96 (m, 2H, H2CCy), 1.86 (s, 3H, CH3C=), 1.71–1.44 (m, 8H, H2CCy), 1.21 ppm (d, JHH = 6.8, 12 H, CHCH3); MS (EI): m/z: 326 [M+H]+.
Representative procedure for the synthesis of alkenyl iminium salts 3: A solution of HCl in Et2O (1.54 mL, 2.0M, 3.1 mmol) was added to a solution of alkenyl aldimine 2a (1.00g, 3.1 mmol) in hexane (10 mL) at −78 °C. Precipitation of a white powder was immediately observed. After 15 minutes the mixture was warmed to room temperature and stirring was continued for an additional 15 minutes. Filtration of the white precipitate, washing with hexanes (2 × 10 mL), and drying under vacuum afforded the alkenyl iminium salt 3a in 92% yield. M.p. 83°C (decomp); 13C{1H} NMR (75.1 MHz, CDCl3, 25 °C): δ = 189.8 (NH=CH), 143.0 (Cortho), 140.5 (C=CH2), 135.4 (Cipso), 130.4 (Cpara), 124.5 (Cmeta), 117.0 (=CH2), 46.7 (CCy), 45.7 (CH2), 34.0 (H2CCy), 28.7 (CHCH3), 25.1 (CH3C=), 25.0 (H2CCy), 24.0 (CHCH3), 22.6ppm (H2CCy); 1H NMR (300.0 MHz, CDCl3, 25°C): δ = 15.50 (s, 1H, NH), 7.98 (s, 1H, CH=N), 7.37 (t, JHH = 8.1 Hz, 1H, Hpara), 7.22 (d, JHH = 8.1 Hz, 2H, Hmeta), 4.99 (s, 1H, C=CH2), 4.82 (s, 1H, C=CH2), 2.99 (sept, JHH = 6.8Hz, 2H, CHCH3), 2.68 (s, 2H, CH2), 2.42 (m, 2H, H2CCy), 1.90 (m, 2H, H2CCy), 1.84 (s, 3H, CH3C=), 1.73 (m, 2H, H2CCy), 1.58 (m, 4H, H2CCy), 1.24 ppm (d, JHH = 6.8 Hz, 12H, CHCH3); MS (FAB): m/z: 326 [M]+.
Representative procedure for the hydroiminiumation reaction leading to 4: A solution of alkenyl iminium salt 3a (1.00 g, 2.8 mmol) in acetonitrile (10 mL) in a tube sealed by a teflon stopcock was heated at 50 °C for 18 h. The volatiles were removed under vacuum to afford 4a as a white powder in 88% yield. Alternatively, a solution of HCl in Et2O (3.08 mL, 2.0M, 6.2 mmol) was added to a solution of alkenyl aldimine 2a (1.00g, 3.1 mmol) in acetonitrile (10mL) at − 78 °C. The solution was warmed to room temperature and sealed with a teflon stopcock, then heated at 50 °C for 18 h. The volatiles were removed under vacuum to afford 4a as a white powder in 88% yield. M.p. 168°C; 13CfH) NMR (75.1 MHz, CDCl3,25°C): δ = 193.0 (N=CH), 144.6 (Cortho), 131.9 (Cpara), 129.0 (Cipso), 125.4 (Cmeta), 82.9 (CCH3), 53.6 (CCy), 45.6 (CH2), 33.8 (H2CCy), 30.0 (CHCH3), 29.1 (CH3), 26.8 (CH3), 24.2 (H2CCy), 22.3 (CH3), 21.3 ppm (H2CCy); 1H NMR (300.0 MHz, CDCl3, 25 °C): δ = 10.69 (s, 1H, CH=N), 7.42 (t, JHH = 7.8 Hz, 1H, Hpara, 7.23 (d, JHH = 7.8 Hz, 2H, Hmeta), 2.57 (sept, JHH = 6.7Hz, 2H, CHCH3), 2.37 (s, 2H, CH2), 1.80–1.34 (m, 10H, H2CCy), 1.47 (s, 6H, CCH3), 1.25 (d, JHH = 6.7 Hz, 6H, CHCH3), 1.13 ppm (d, JHH = 6.7 Hz, 6H, CHCH3); MS (FAB): m/z: 326 [M]+. All spectroscopic data are comparable to those observed for the corresponding triflate salt. [12a]
Footnotes
We are grateful to the NIH (R01 CM 68825) and Rhodia for financial, support of this work.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.O’Hagan D. Nat Prod Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see: Matsunaga S. J Synth Org Chem Jpn. 2006;64:778–779.Hultzsch KC. Adv Synth Catal. 2005;347:367–391.Alonso F, Beletskaya IP, Yus M. Chem Rev. 2004;104:3079–3159. doi: 10.1021/cr0201068.Roesky PW, Muller TE. Angew Chem. 2003;775:2812–2814.Angew Chem Int Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637.Pohlki F, Doye S. Chem Soc Rev. 2003;32:104–114. doi: 10.1039/b200386b.Muller T, Beller M. Chem Rev. 1998;98:675–703. doi: 10.1021/cr960433d.
- 3.Hartung CG, Breindl C, Tillack A, Beller M. Tetrahedron. 2000;56:5157–5162. [Google Scholar]
- 4.For reviews, see: Roesky PW. Z Anorg Allg Chem. 2006;652:1918–1926.Odom AL. Dalton Trans. 2005:225–233. doi: 10.1039/b415701j.Hazari N, Mountford P. Acc Chem Res. 2005;38:839–849. doi: 10.1021/ar030244z.Bytschkov I, Doye S. Eur J Org Chem. 2003:935–946.
- 5.For recent examples, see: Zhang J, Yang CG, He C. J Am Chem Soc. 2006;128:1798–1799. doi: 10.1021/ja053864z.Komeyama K, Morimoto T, Takaki K. Angew Chem. 2006;118:3004–3007. doi: 10.1002/anie.200503789.Angew Chem Int Ed. 2006;45:2938–2941. doi: 10.1002/anie.200503789.Michael FE, Cochran BM. J Am Chem Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h.Johns AM, Sakai N, Ridder A, Hartwig JF. J Am Chem Soc. 2006;128:9306–9307. doi: 10.1021/ja062773e.Takemiya A, Hartwig JF. J Am Chem Soc. 2006;128:6042–6043. doi: 10.1021/ja058299e.Meyer N, Lohnwitz K, Zulys A, Roesky PW, Dochnahl M, Blechert S. Organometallics. 2006;25:3730–3734.Liu XY, Li CH, Che CM. Org Lett. 2006;8:2707–2710. doi: 10.1021/ol060719x.Zulys A, Dochnahl M, Hollmann D, Lohnwitz K, Herrmann JS, Roesky PW, Blechert S. Angew Chem. 2005;777:7972–7976.Angew Chem Int Ed. 2005;44:7794–7798. doi: 10.1002/anie.200502006.
- 6.a) Gribkov DV, Hultzsch KC, Hampel F. J Am Chem Soc. 2006;128:3748–3759. doi: 10.1021/ja058287t. [DOI] [PubMed] [Google Scholar]; b) Riegert D, Collin J, Meddour A, Schulz E, Trifonov A. J Org Chem. 2006;77:2514–2517. doi: 10.1021/jo052322x. [DOI] [PubMed] [Google Scholar]; c) Arnea E, Eisen MS. Coord Chem Rev. 2006;250:855–859. [Google Scholar]; d) Hong S, Marks TJ. Ace Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 7.a) Schlummer B, Hartwig JF. Org Lett. 2002;4:1471–1474. doi: 10.1021/ol025659j. [DOI] [PubMed] [Google Scholar]; b) Motokura K, Nakaagiri N, Mori K, Mizugaki T, Ebitani K, Jitsukawa K, Kaneda K. Org Lett. 2006;8:4617–4620. doi: 10.1021/ol0619821. [DOI] [PubMed] [Google Scholar]
- 8.a) Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org Lett. 2006;8:4179–4182. doi: 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]; b) Anderson LL, Arnold J, Bergman RG. J Am Chem Soc. 2005;727:14542–14543. doi: 10.1021/ja053700i. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lapis AAM, DaSilveira Neto BA, Scholten JD, Nachtigall FA, Eberlin MN, Dupont J. Tetrahedron Lett. 2006;47:6775–6779. [Google Scholar]; d) Li ZG, Zhang JL, Brouwer C, Yang CG, Reich NW, He C. Org Lett. 2006;8:4175–4178. doi: 10.1021/ol0610035. [DOI] [PubMed] [Google Scholar]
- 9.a) Nicolaou KC, Baran PS, Baran YL, Zhong YL, Sugita K. J Am Chem Soc. 2002;724:2212–2220. doi: 10.1021/ja012124x. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Baran PS, Zhong YL, Barluenga S, Hunt KW, Kranich R, Vega JA. J Am Chem Soc. 2002;724:2233–2244. doi: 10.1021/ja012126h. [DOI] [PubMed] [Google Scholar]; c) Nicolaou KC, Zhong YL, Baran PS. Angew Chem. 2000;772:636–639. [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:622–625. [PubMed] [Google Scholar]; d) Nicolaou KC, Zhong YL, Baran PS. Angew Chem. 2000;772:639–642. [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:625–628. doi: 10.1002/(sici)1521-3773(20000204)39:3<625::aid-anie625>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 10.a) Mangelinckx S, Giubellina N, De Kimpe N. Chem Rev. 2004;104:2353–2399. doi: 10.1021/cr020084p. [DOI] [PubMed] [Google Scholar]; b) De Kimpe N, De Smaele D. J Heterocycl Chem. 2000;37:607–614. [Google Scholar]
- 11.De Kimpe N, Boelens M. J Chem Soc Chem Commun. 1993:916–918. [Google Scholar]
- 12.a) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew Chem. 2005;777:5851–5855. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:5705–5709. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lavallo V, Canac Y, Dehope A, Donnadieu B, Bertrand G. Angew Chem. 2005;777:7402–7405. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:7236–7239. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For reviews on NHCs: Hahn FE. Angew Chem. 2006;118:1374–1378.Angew Chem Int Ed. 2006;45:1348–1352. doi: 10.1002/anie.200503858.Scott NM, Nolan SP. Eur J Inorg Chem. 2005:1815–1828.Kuhn N, Al-Sheikh A. Coord Chem Rev. 2005;249:829–857.Peris E, Crabtree RH. Coord Chem Rev. 2004;248:2239–2246.Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247–2273.Cesar V, Bellemin-Laponnaz S, Gade LH. Chem Soc Rev. 2004;33:619–636. doi: 10.1039/b406802p.Enders D, Balensiefer T. Acc Chem Res. 2004;37:534. doi: 10.1021/ar030050j.Herrmann WA. Angew Chem. 2002;774:1342–1363. doi: 10.1002/1521-3773(20020415)41:8<1363::aid-anie1363>3.0.co;2-g.Angew Chem Int Ed. 2002;47:1290–1309.Bourissou D, Guerret O, Gabbaï FP, Bertrand G. Chem Rev. 2000;100:39–92. doi: 10.1021/cr940472u.Arduengo AJ., III Ace Chem Res. 1999;32:913–921.
- 14.CCDC-626900 (3a), CCDC-626901 (3g), and CCDC-626902 (4g) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 15.One of the reviewers suggested that the preference for five-membered versus six-membered heterocycles could be in correlation with Baldwin’s rules: Johnson CD. Acc Chem Res. 1993;26:476–482.