

Abstract
We have shown previously that oxidized phospholipids (OxPLs), known to accumulate in atherosclerotic vessels, stimulate angiogenesis via induction of autocrine mediators, such as vascular endothelial growth factor (VEGF). We now address the pathways mediating up-regulation of VEGF in human endothelial cells treated with OxPLs. Analysis of structure-function relationship using individual species of OxPLs demonstrated a close relation between induction of VEGF and activation of the unfolded protein response (UPR). Inducers of UPR up-regulated VEGF, whereas inhibition of UPR by chemical chaperones or knock-down of cochaperone HTJ-1 inhibited elevation of VEGF mRNA induced by OxPLs. OxPLs induced protein expression of activating transcription factor-4 (ATF4), an important effector of UPR. Expression levels of VEGF in OxPL-treated cells strongly correlated with induction of the ATF4 target genes ATF3 and TRB3. Knocking down ATF4 was paralleled by loss of VEGF induction by OxPLs. Chromatin immunoprecipitation demonstrated that OxPLs stimulated binding of ATF4 to a regulatory site in the VEGFA gene. Taken together, these data characterize UPR and more specifically its ATF4 branch as an important mechanism mediating up-regulation of VEGF by OxPLs, and allow hypothesizing that the UPR cascade might play a role in pathologic angiogenesis in atherosclerotic plaques.
Introduction
A key feature of atherosclerosis is accumulation of insoluble lipid deposits within the arterial wall. Oxidized phospholipids (OxPLs) were characterized by several laboratories as a common lipid component of atheroma.1–4 OxPLs accumulate within the arterial wall as a consequence of oxidative stress and pathologic cellular lipid uptake, but it is increasingly recognized that they are not only markers but also active mediators of atherogenesis. This view is supported by numerous reports on the ability of OxPLs to induce in vitro and in vivo a number of cellular atherogenic effects, such as stimulation of monocyte-endothelial cell (EC) interaction, production of chemokines, and procoagulant shift in endothelium.5 OxPLs containing hydroperoxide groups are also capable of propagating peroxidation chain reactions, further enhancing oxidative stress within the arterial wall. Furthermore, it has been shown that OxPLs induce endoplasmic reticulum (ER) stress leading to the unfolded protein response (UPR).6 On the other hand, it is becoming increasingly clear that OxPLs are not solely proatherogenic stress agents but also can induce antiinflammatory and tissue-protective effects, such as up-regulation of the antioxidant and antiinflammatory protein heme oxygenase-1, inhibition of inflammation induced by bacterial products via Toll-like receptors,7,8 and enhancement of the lung EC barrier, thus protecting the endothelial lining from agents inducing edema.9,10 In summary, OxPLs represent a group of lipid mediators inducing a rapidly growing list of cellular effects relevant to acute and chronic inflammation.
One explanation for the functional variability of the effects of OxPLs is the complexity of lipid peroxidation reactions generating a wide spectrum of full-length or fragmented derivatives of polyunsaturated fatty acids containing various numbers and combinations of hydroperoxides, aldehydes, carboxyls, cyclopentenons, and other groups. Furthermore, phospholipases can cleave oxidatively modified residues, thus forming unesterified oxidation products and lysophospholipids. Therefore, it is not surprising that phospholipid oxidation products have been suggested to activate several types of signaling receptors, including PAF receptor, prostaglandin EP2 receptor, PPARs, and TLRs.11–14 Inside the cell, OxPLs activate variable signaling mechanisms, including cAMP- and Ca2+-dependent pathways, MAP kinase cascades, small GTPases R-Ras, Rac and Cdc42, and other signaling molecules.5,15,16 Thus, OxPLs represent a heterogeneous group of lipid mediators demonstrating variable cellular effects mediated by a number of signal transduction mechanisms.
Previously, we have found that OxPLs induce angiogenic effects in several in vitro and in vivo models.17 We identified autocrine loops mediating the angiogenic action of OxPLs, such as vascular endothelial growth factor (VEGF), interleukin-8 (IL-8), and cyclooxygenase-2 (COX-2)-derived prostaglandins.17 In this work, we sought to examine the mechanisms mediating VEGF induction by OxPLs. Furthermore, we analyzed structural characteristics of OxPLs responsible for their angiogenic activity. We show that the activating transcription factor-4 (ATF4) branch of the UPR is a key mechanism that mediates OxPL-induced up-regulation of VEGF. Furthermore, we demonstrate that the oxidized sn-2 residue is the most important structural characteristic required for VEGF induction and UPR activation. These data provide further insights into the mechanisms of the pathogenic activity of lipids accumulated in atheroma.
Methods
Materials
Thapsigargin, tunicamycin, brefeldin A, D,L-homocysteine, dithiothreitol, sodium 4-phenylbutyrate, 1-O-hexadecyl-2-acetyl-sn-glycero-3-phosphocholine (platelet activating factor, PAF), lipopolysaccharide from Escherichia coli serotype 055:B5, and actinomycin D were purchased from Sigma-Aldrich (Vienna, Austria). Tiron was from Fluka (Vienna, Austria). Salubrinal and rosiglitazone were from Alexis Biochemicals (Lausen, Switzerland). WY-14643, gingkolide B, and CV-3988 were from Biomol (Hamburg, Germany); 8-iso-prostaglandin E2 was purchased from Cayman Chemical (Ann Arbor, MI). Anti-HIF-1α was from BD Biosciences (Vienna, Austria). Anti-EGR-1 was from Santa Cruz Biotechnology (Santa Cruz, CA), anti-actin from DakoCytomation (Vienna, Austria). Antibodies to total and phosphorylated eIF2α were purchased from Cell Signaling Technology (Frankfurt am Main, Germany). Antibodies against C-terminus of ATF4 and an epitope within amino acids 1 to 290 were obtained from Santa Cruz Biotechnology. Organic solvents and other chemicals were of analytical grade.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated as described18 and cultured at 37°C and 5% CO2 in medium 199 containing 20% fetal calf serum (FCS), 1 U/mL heparin, 50 μg/mL bovine EC growth supplement (Technoclone, Vienna, Austria), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Experiments were performed using cells up to passage 5. In all experiments, HUVECs were stimulated with lipids and other agonists in medium 199 containing 2% FCS. Cultivation and stimulation of human aortic endothelial cells (HAECs) was performed as described previously.6
Lipids
Synthetic 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoglycerol (PAPG), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphate, 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoserine, 1-palmitoyl-2-hydroxy-sn-3-glycero-phosphocholine (lysoPC), and 1-palmitoyl-2-linoleoyl-sn-3-glycerophosphocholine were purchased from Avanti Polar Lipids (Alabaster, AL). Arachidonic acid was from Sigma-Aldrich. Dry lipids were oxidized by exposure to air until approximately 80% of the lipid was oxidized. Oxidized lipids were dissolved in chloroform, purged with argon and stored at −70°C. Oxidation was monitored by thin-layer chromatography and electrospray ionization-mass spectrometry.4 1-Palmitoyl-2-(5,5′-dimethoxyvaleroyl)-sn-glycero-3-phosphocholine (acetal-POVPC), 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC), and 1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphocholine were synthesized from lysoPC according to the previously published protocol,4 except for using dicyclohexylcarbodiimide immobilized on polymer beads (Novabiochem, San Diego, CA). 1-Palmitoyl-2-(5,6-epoxy isoprostane E2)-sn-glycero-3-phosphocholine (isoprostane-PC)-enriched fraction was isolated from OxPAPC by high performance liquid chromatography purification on a Adsorbosphere Si 100 column (5 μm, 250 × 10 mm, Alltech, Hamburg, Germany) using isocratic elution with a mixture of acetonitrile/methanol/water (77:8:15, vol/vol) supplemented with 0.02% (vol/vol) of triethylamine at the flow rate of 3 mL/min. A mixture of 1-palmitoyl-2-(12-hydroperoxyeicosa-5,8,10,14-tetraenoyl)-sn-glycero-3-phosphocholine and 1-palmitoyl-2-(15-hydroperoxyeicosa-5,8,11,13-tertraenoyl)-sn-glycero-3-phosphocholine (PAPC-OOH) was produced from PAPC by enzymatic reaction with a soybean lipoxygenase.19 PAPC-OOH was purified on solid phase extraction cartridge using a previously published protocol.20 Measurement of lipid peroxides was performed using a ferric thiocyanate method.21 PAPC hydroxide (PAPC-OH) was obtained from PAPC-OOH by reduction with triphenylphosphine.17 Concentration of phospholipids was determined by phosphorus assay.22 Before adding to cells, the lipids were resuspended by vigorous vortexing in medium 199 supplemented with 2% FCS.
Promoter-reporter assay
HUVECs grown in 6-well dishes were cotransfected with 1.5 μg/well of pGL3 promoter plasmid (Promega, Madison, WI) containing 4 hypoxia response elements of the Epo gene23 and 0.2 μg pRL-SV40 (for normalization of transfection efficiency) using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA) as previously described by us.24 Twenty-four hours after transfection, the medium was changed to M199/2% FCS containing 130 μM OxPAPC. After overnight incubation, cells were lysed and luciferase activity was measured using a dual luciferase assay system (Promega). The data are expressed as firefly luciferase activity normalized to the Renilla luciferase activity.
mRNA quantification by real-time reverse transcription PCR
Reverse transcription followed by quantitative PCR (RT-qPCR) was performed as described by us previously.25 Briefly, RNA was isolated using Trizol reagent (Invitrogen) according to the manufacturer's protocol; 1 μg total RNA was taken for cDNA synthesis using the GeneAmp RNA PCR kit and oligo d(T)16 primers (Applied Biosystems, Foster City, CA). Real-time PCR was performed using a LightCycler instrument (Roche Diagnostics, Vienna, Austria) and FastStart SYBR Green Master Mix (Roche Diagnostics). Sequences of primers are available on request.
mRNA stability assay
Actinomycin D (5 μg/mL) was added to HUVECs preincubated for 3 hours with or without 130 μM OxPAPC. Triplicate samples were harvested at the time points indicated in Figure 1A, and combined for further analysis. RNA isolation, reverse transcription, and quantitative PCR were performed as described in “mRNA quantification by real time reverse transcription PCR.” Levels of COX-2 and VEGF mRNAs were normalized to the levels of β2-microglobulin mRNA. The amount of mRNA expressed immediately before the addition of actinomycin D was set as 100%.
Figure 1.

OxPLs do not influence VEGF mRNA stability and do not up-regulate HIF-1α protein or HIF-1–dependent transcription. (A) HUVECs were incubated in medium 199 containing 2% FCS with or without 130 μM OxPAPC. Actinomycin D was added after 4 hours to stop transcription. After indicated time intervals, the samples were collected and RNA was extracted, reverse transcribed, and analyzed by RT-qPCR for expression of VEGF or COX-2 mRNA. (B) HUVECs were incubated in medium 199 containing 2% FCS with or without 130 μM OxPAPC or 30 mM metal chelator Tiron. At the indicated time points, the cells were scraped into Laemmli sample buffer and analyzed by Western blotting. Staining of the same samples for HIF-1α or EGR-1 proteins is shown. (C) HUVECs grown in 6-well dishes were cotransfected with firefly luciferase promoter-reporter driven by hypoxia response elements and constitutively active Renilla luciferase. After 24 hours, the medium was changed to 2% FCS containing indicated concentrations of OxPAPC (100 μg/mL of OxPAPC = 130 μmol/L). After an overnight incubation, the cells were scraped and activities of both luciferases measured using a dual luciferase assay system. The data represent a ratio of firefly luciferase activity to that of Renilla luciferase. Error bars represent SD.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) analysis was performed using an EZ-Chip kit (Upstate Biotechnology, Charlottesville, VA) according to the manufacturer's instructions. HUVECs or HeLa cells were incubated for 3 hours with or without OxPAPC (130 μM) in M199 or DMEM media, respectively, containing 2% FCS. Thereafter, formaldehyde was added (final concentration, 1%) and cells further incubated at room temperature. DNA-protein cross-linking was stopped by the addition of glycine (to 0.125 mM) after 8 and 10 minutes of incubation for HUVECs and HeLa cells, respectively. Cell extracts were sonicated using Bandelin Sonopuls sonicator (Bandelin Electronic, Berlin, Germany; HUVECs: 3 bursts × 10 seconds at 30% power; HeLa cells: 5 bursts × 10 seconds). Extracts from 107 cells were incubated overnight with antibodies against ATF4 or nonimmune rabbit IgG provided in EZ-Chip kit. One percent of extract volume was removed before immunoprecipitation and served as input control. DNA fragments from immunoprecipitated complexes and inputs were released by heating at 65°C overnight and purified according to the manufacturer's protocol. Purified immunoprecipitated and input DNAs were analyzed by PCR. The ATF4-binding sites and their locations within the VEGF promoter were described previously.26 For amplification of gene regions containing ATF4-binding sites, the following primers were used: 5′-TTGCCTCCTGGGTTTAAGTG-3′ and 5′-GAAACAGCACTGGGCAAAA-3′ for VEGF “CHOP1” site, 5′-GCGGAGCCGATTACATCA-3′and 5′-ACTGGGCAGAGGTAGGGTCT-3′ for VEGF “AsnSyn” site, and 5′-AACAAAAGAGCTCCTCCTTGC-3′and 5′-AGGGATGTGGACAGCTTGAC-3′ for the NSRE-1 site in asparagine synthetase gene.27 PCR products were resolved on agarose gels and visualized by ethidium bromide staining.
siRNA-mediated gene knock-down
siRNA targeting HTJ1, ATF4 siRNA, and control siRNA were obtained from Ambion (Brunn am Gebirge, Austria). HUVECs were transfected with 100 nmol/L of siRNA using RNAiFect Transfection Reagent (QIAGEN, Vienna, Austria) according to the manufacturer's instructions. Forty-eight hours after transfection, the cells were treated with OxPAPC. Efficiency of ATF4 and HTJ1 knock-down was confirmed by RT-qPCR analysis of their mRNAs.
Western blotting
Cells were lysed in Laemmli buffer and applied onto SDS-polyacrylamide gels. Proteins were transferred onto Immobilon-P (Millipore, Vienna, Austria) membranes by electroblotting. After blocking in phosphate-buffered saline containing 5% dry milk, the membranes were incubated with polyclonal rabbit antibodies against ATF4, HIF-1α, EGR-1, or actin. Bound primary antibodies were detected with antirabbit IgG conjugated with peroxidase and visualized using the LumiGLO chemiluminescent substrate (Cell Signaling Technology) in combination with autoradiography or chemiluminescent imager FluorChem HD2 (Alpha Innotech, San Leandro, CA).
Statistical analysis
All results are expressed as mean values plus or minus SD from triplicate or quadruplicate measurements performed in 2 to 4 independent experiments producing similar results. Where indicated, the data were analyzed by 2-tailed Student t test. A P value less than .05 was regarded as significant.
Results
Effects of OxPLs on VEGF mRNA stability
Regulation of VEGF expression is complex and occurs at transcriptional but also posttranscriptional levels.28 Therefore, we first tested whether OxPLs regulate stability of VEGF mRNA. OxPLs did not significantly influence kinetics of VEGF mRNA degradation (Figure 1A). In the same cell preparations, OxPLs significantly enhanced stability of COX-2 mRNA (Figure 1A). These data suggest that OxPLs regulate VEGF mRNA expression at the level of gene transcription.
Effects of OxPLs on HIF-1
Expression of VEGF is regulated by a number of transcription factors, the most important being HIF-1.29 HIF-1 mediates induction of VEGF by hypoxia, generally regarded as the universal angiogenic stimulus in particular being pivotal in neovascularization of atherosclerotic plaques.30 In addition to hypoxia, many other stimuli relevant to atherosclerosis, including oxidized LDL and reactive oxygen species, also regulate HIF-1–dependent transcription.31–33 Therefore, we analyzed whether HIF-1 plays a role in the induction of VEGF by OxPLs. A key event in the activation of HIF-1–mediated transcription is up-regulation of HIF-1α protein.34 The metal chelator Tiron, known to block degradation of HIF-1α by a mechanism similar to that activated by hypoxia,35 strongly up-regulated HIF-1α protein in HUVECs (Figure 1B). In contrast, OxPLs did not influence the levels of HIF-1α, although they significantly up-regulated EGR-1 previously characterized as an OxPL-inducible gene24 (Figure 1B). Furthermore, OxPLs did not activate luciferase promoter-reporter construct driven by hypoxia response elements known to bind HIF-1 (Figure 1C). In summary, these data indicate that induction of VEGF by OxPLs is unlikely to be mediated via a HIF-1/hypoxia response element-dependent transcription.
Analysis of structure-function relationship of VEGF induction by phospholipids
To clarify receptor and signaling mechanisms activated by OxPLs, we further analyzed structural characteristics of oxidized sn-2 residues important for up-regulation of VEGF. To this end, we compared the VEGF-inducing activity of several molecular species present in OxPAPC (Figure 2A). Species with oxidatively fragmented sn-2 residues up-regulated VEGF mRNA essentially independently of the type of ω-terminal group, such as aldehyde in POVPC, carboxyl in 1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphocholine, or dimethyl acetal in POVPC-acetal. We also observed induction of VEGF by OxPLs containing diverse full-length oxygenated sn-2 residues, such as hydroperoxides, their reduced hydroxy derivatives, or OxPLs containing isoprostanes. Altogether, the data of Figure 2A demonstrate nonstringent structural requirements for the oxidized sn-2 residue necessary for VEGF induction. However, the presence of sn-2 residue is critically important for VEGF induction because lysophospholipid was dramatically less active compared with OxPLs (Figure 2B).
Figure 2.

Lipid structural specificity of OxPL-induced up-regulation of VEGF mRNA. HUVECs were stimulated for 6 hours with indicated lipids resuspended in medium 199 containing 2% FCS. The incubation was terminated by Trizol, followed by RNA extraction and VEGF mRNA quantification by RT-qPCR. The expression levels of VEGF were normalized to those of β2-microglobulin mRNA. (A) HUVECs were stimulated with 130 μM of OxPAPC or individual OxPLs. (B) HUVECs were treated with increasing concentrations of OxPAPC or lysoPC. (C) The stimulation of HUVECs was performed by increasing concentrations of oxidized arachidonic acid (OxAA). (Inset) Results of HUVEC stimulation with 130 μM of OxPAPC, oxidized (OxAA) or unoxidized arachidonic acid (AA). Error bars represent SD.
The data described in the previous paragraph characterized oxidized sn-2 residues as key determinants of the activity of OxPLs. In the following experiments, we asked the question whether oxidized residues are active only when esterified or can stimulate cells as free unesterified molecules. To test this possibility, we treated HUVECs with unesterified arachidonic acid (AA) oxidized according to the same protocol as phospholipids. Unesterified OxAA stimulated production of VEGF acting in molar concentrations comparable with those of OxPLs (Figure 2C). To rule out secondary effects of prostanoids formed by cells from unoxidized AA present in the OxAA preparation, we treated HUVECs with pure unoxidized AA. Unoxidized AA was inactive (Figure 2C, inset), thus demonstrating that delivery of substrate for prostaglandin synthesis is not critical for stimulation of VEGF expression by OxAA or OxPLs. In summary, these data show that OxAA induces VEGF when added either in phospholipid-esterified or free forms.
Analysis of the involvement of PAF-, EP2-, PPAR-, Toll-like, and lysophospholipid receptors
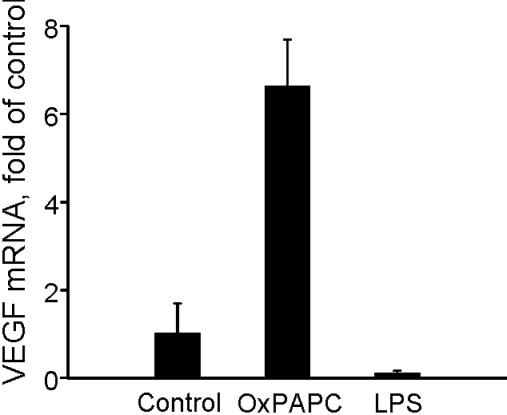
The data on the structure-function relationship of VEGF up-regulation by OxPLs allowed us to address the question on the role of previously characterized or putative receptors for OxPLs. We found that structural requirements for the VEGF-inducing activity did not correspond to the known ligand selectivity of PAF, prostaglandin EP2, or lysophospholipids receptors (Document S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Furthermore, addition of PAF did not influence the levels of VEGF mRNA, and 2 PAF-receptor antagonists did not inhibit OxPL-induced up-regulation of VEGF (Figure S1A,B). Similarly, iso-prostaglandin E2, which is known to be present in OxPAPC and is on the other hand a putative EP2-receptor ligand,36 did not influence the levels of VEGF mRNA (Figure S2). In addition, lipopolysaccharide did not elevate expression of VEGF in HUVECs (Figure S3), thus ruling out a role for Toll-like receptor 4. We also observed a lack of VEGF induction by PPARα or PPARγ agonists (Figure S4). In summary, our data do not support any role for previously characterized or hypothesized OxPL receptors in VEGF induction, thus necessitating search for additional receptor-dependent and/or receptor-independent signaling mechanisms mediating the effects of OxPLs on VEGF expression.
Role of UPR activation in OxPL-induced up-regulation of VEGF
It has been shown that lipid-induced perturbations of cell membranes resulting from accumulation of free cholesterol or gangliosides37–39 can induce ER stress followed by a cellular adaptation process known as UPR.40,41 More recently, it has been shown that OxPLs induce ER stress and UPR in ECs.6 Because the VEGFA promoter contains functional binding sites for transcription factor ATF4,26 which is one of the important effectors of UPR,42 we hypothesized that OxPL-induced UPR may be a mechanism that activates VEGF transcription. In support of this hypothesis, we found that chemically different inducers of ER stress such as dithiotreitol, tunicamycin, thapsigargin, and brefeldin A all up-regulated VEGF mRNA (Figure 3A). On a molar basis, OxPLs were 2 orders of magnitude more potent in inducing VEGF compared with another atherosclerosis risk factor homocysteine previously characterized as a UPR inducer43 (Figure 3B). To demonstrate the role of UPR in activation of VEGF expression by OxPLs, we used chemical chaperones, which are low-molecular-weight compounds stabilizing protein structure and thus inhibiting activation of UPR by a variety of ER-stress inducers.44 We found that 4-phenylbutyrate45 (PBA) inhibited OxPL- and tunicamycin-induced up-regulation of VEGF (Figure 3C), as well as UPR genes ATF3 and TRB3(data not shown). Furthermore, we found that knocking down the cochaperone HTJ-1 that stimulates the ATPase activity of GRP78/BIP necessary for stabilizing its complexes with unfolded substrates,46,47 strongly inhibited VEGF up-regulation by OxPLs (Figure 3D). Taken together, these data strongly suggest that UPR plays an important role in activation of VEGF expression by OxPLs.
Figure 3.

Unfolded protein response up-regulates VEGF mRNA. HUVECs were treated for 6 hours in medium 199/2% FCS containing (A) 130 μM OxPAPC, 1 mM dithiothreitol (DTT), 3 μg/mL tunicamycin, 1 μg/mL thapsigargin, or 6 μg/mL brefeldin A, or (B) with increasing concentrations of homocysteine dissolved in medium 199 containing 2% FCS. (C) HUVECs were preincubated overnight with or without 2.5 mM sodium PBA in full growth medium. On the next day, the cells were stimulated for 6 hours with 130 μM OxPAPC or 3 μg/mL tunicamycin in medium 199 containing 2% FCS with or without PBA. After mRNA isolation, levels of mRNA encoding for VEGF were quantified by RT-qPCR. (D) HUVECs were transfected with siRNA against HTJ1 or control siRNA. Forty-eight hours later, the cells were stimulated for 6 hours with 130 μM OxPAPC in medium 199 containing 2% FCS. The incubations were terminated by Trizol, followed by quantification of VEGF mRNA. The data are normalized to the levels of β2-microglobulin mRNA. Error bars represent SD.
Analysis of structure-function relationship of UPR induction by phospholipids
As an additional approach to demonstrate that the induction of VEGF by OxPLs was mediated by UPR, we determined the lipid specificity of OxPL-induced UPR using ATF4 and its downstream genes ATF3 and TRB3 as readouts.48,49 We found that different classes of OxPLs stimulated expression of ATF4 protein and ATF3 mRNA (Figure 4A,B). Indeed, we have observed previously a similar dependence on the structure of polar head groups for the activation of VEGF expression.17 ATF3 expression was induced by both fragmented and full-length species of OxPLs, and the profile of activity was similar to that of VEGF induction (Figures 2A,4C). Similarly to the up-regulation of VEGF, the lack of sn-2 residue dramatically decreased up-regulation of ATF3 (Figures 2B,4D). OxAA but not native AA strongly induced ATF3 (Figures 2C,4E). To summarize, we found that oxidized sn-2 residues are key structural determinants of UPR activation by OxPLs and that structural requirements for activation of UPR by different OxPLs are very similar to the regulation of VEGF expression.
Figure 4.

Lipid structural specificity of OxPL-stimulated induction of UPR genes. HUVECs were stimulated with lipids in medium 199 containing 2% FCS. All data obtained by RT-qPCR are normalized to the levels of β2-microglobulin mRNA. (A) HUVECs were incubated with 130 μM OxPLs containing palmitic and arachidonic acid residues: phosphatidylcholine (PC), phosphatidylglycerol (PG), phosphatidic acid (PA), or phosphatidylserine (PS). Control cells were incubated with the medium containing no lipids. After 4 hours, the cells were scraped into Laemmli buffer and analyzed by Western blotting for ATF4. (B) HUVECs were treated with oxidized or unoxidized phospholipids for 6 hours and then processed for quantification of ATF3 mRNA by RT-qPCR. (C) HUVECs were incubated with individual OxPLs (130 μM each) for 6 hours. (D) HUVECs were incubated for 6 hours with increasing concentrations of OxPAPC or lysoPC. (E) HUVECs were treated with the indicated concentrations of oxidized arachidonic acid (OxAA). (Inset) Data obtained after treatment of HUVECs for 6 hours with 130 μM OxPAPC or oxidized (OxAA) and unoxidized (AA) arachidonic acid. Error bars represent SD.
Role of ATF4 in OxPL-induced up-regulation of VEGF
To address the involvement of the ATF4 branch of the UPR in VEGF up-regulation, we analyzed correlation between the expression levels of VEGF and ATF4 downstream genes. We found strong correlation between induction of VEGF, ATF3, and TRB3 in samples treated with a variety of different OxPLs (Figure 5A,B), suggesting a possibility of common mechanisms for the regulation of these genes. Close parallels between the lipid specificities of VEGF and ATF3/TRB3 induction allow to hypothesize that the ATF4 branch of UPR represents a mechanism mediating induction of VEGF by OxPLs. In support of this notion, we found that knocking down ATF4 by siRNA (Figure S5) significantly inhibited OxPL-induced increase in VEGF (Figure 5C). Similar inhibitory effect of ATF4 siRNA on OxPL-induced up-regulation of VEGF was also reproducibly observed in HAECs (Figure 5D). Because the selective translation of ATF4 critically depends on the phosphorylation state of eIF2α, we tested whether modulation of eIF2α phosphorylation can regulate VEGF expression induced by OxPLs. Indeed, production of VEGF mRNA was stimulated by salubrinal (Figure 5E) known to inhibit dephosphorylation of eIF2α, thus leading to enhanced translation of ATF4.50 These results further implicate the eIF2α-ATF4 pathway as a mechanism of VEGF induction by OxPLs. Time kinetic measurements showed that phosphorylation state of eIF2α and levels of ATF4 protein started to increase within 1 hour of incubation with OxPLs and reached steady-state levels by 2 hours (Figure 5F,G), thus preceding the onset of VEGF mRNA elevation that, as we have shown previously, was only marginal after 2 hours.17 This time course is compatible with the possibility that transcription of VEGF is directly activated by ATF4. This notion was further supported by the data of ChIP showing that OxPLs stimulated binding of ATF4 to the regulatory “AsnSyn” site26 in the VEGFA gene (Figure 5H top panel). The importance of the “AsnSyn” site for activation of VEGF transcription by ATF4 was documented previously.26 A specific PCR product was present in samples immunoprecipitated with anti-ATF4 (Figure 5H all panels). The same PCR product was observed in preparations of cells treated with UPR activator tunicamycin (Figure 5H top panel). In addition, OxPAPC and tunicamycin stimulated formation of a complex between ATF4 and a functionally important NSRE-1 site in the promoter of asparagine synthetase27 (Figure 5H middle panel). In contrast, we observed no binding to another predicted ATF4 site in the VEGFA promoter previously called “CHOP1” site26 (Figure 5H bottom panel). The “CHOP1” site in the VEGF promoter was characterized as nonfunctional,26 thus providing an additional control for the specificity of the ChIP procedure. The effect of OxPAPC on ATF4/DNA interaction was not restricted to ECs as illustrated by immunoprecipitation by anti-ATF4 of the “AsnSyn”-containing VEGFA gene fragment from cervical carcinoma HeLa cells stimulated by OxPAPC (Figure S6). In summary, the data of the ChIP analysis support the notion that ATF4 induced by OxPLs directly binds to the “AsnSyn” site in the VEGFA gene and stimulates its transcription.
Figure 5.

Induction of VEGF by OxPLs is mediated by the ATF4 branch of UPR. (A,B) Correlation between the expression levels of VEGF and ATF3 (A) or TRB3 (B) in cells treated with various oxidized and unoxidized phospholipids. The data are collected from multiple experiments using different concentrations and molecular species of phospholipids. In all cases, the incubation was performed for 6 hours. Experiments using inhibitors or siRNA treatment were excluded. (C) HUVECs were transfected with siRNA against ATF4. Forty-eight hours later, the cells were stimulated for 6 hours with 130 μM OxPAPC in medium 199 containing 2% FCS. The incubations were terminated by Trizol, followed by quantification of VEGF mRNA. The data are normalized to the levels of β2-microglobulin mRNA. (D) The experiment was performed as in panel C, but using HAECs. Incubation was performed for 4 hours. (E) HUVECs were pretreated for 30 minutes with 75 μM salubrinal followed by incubation with 130 μM OxPAPC in the presence of salubrinal in medium 199 containing 2% FCS. After 6 hours, the incubation was terminated by Trizol, and the levels of VEGF mRNA were quantified by RT-qPCR. Combined data from 2 independent experiments are shown. (F) HUVECs were incubated with 130 μM OxPAPC in medium 199 containing 2% FCS. After indicated time intervals, the cells were scraped into Laemmli buffer and analyzed by Western blotting for phosphorylated eIF2α. Afterward, the same blots were stained for total eIF2α. Intensity of immunostaining was quantified using chemiluminescent scanner. The normalized signals do not reflect real ratio of phosphoprotein and total protein due to different sensitivities of antibodies and various exposure times. (G) HUVECs were stimulated with OxPAPC (130 μM) resuspended in medium 199 containing 2% FCS. After indicated time intervals, the cells were scraped into Laemmli buffer and analyzed by Western blotting using antibodies against ATF4. (H) HUVECs were treated with OxPAPC (130 μM) or tunicamycin (3 μg/mL) for 3 hours and then processed for ChIP analysis as described in “Chromatin immunoprecipitation.” The figure presents results of ethidium bromide staining of products of PCR amplification of the fragments of VEGF and asparagine synthetase genes. The fragments overlap ATF4-binding sites within the VEGF (“AsnSyn” and “CHOP-1”)26 and asparagin synthetase (NSRE-1)27 genes. The antibodies used for ChIP are indicated on the top. The “input” was obtained by amplification of 1% of initial unfractionated cell extract.
Discussion
The central finding of this study is that OxPLs up-regulate the major angiogenic cytokine VEGF via activation of UPR, and more specifically its ATF4 branch. VEGF is increasingly recognized as having a role in atherogenesis where it acts not only as an inducer of lesion neovascularization, but also as an inflammatory mediator.51,52 Because atherosclerotic vessels are known to contain high concentrations of OxPLs,4 and on the other hand to express UPR, and in particular ATF4 target genes,6,53,54 OxPL-induced UPR is likely to be a mechanism partially responsible for increased concentrations of VEGF found in atheroma.55 Our observations expand the list of proatherogenic effects mediated by UPR, further emphasizing the role of this cellular stress reaction in vascular pathology. It is known that ER stress and UPR are activated by atherosclerosis risk factors such as homocysteine and cholesterol.37,43 Furthermore, previous findings demonstrated that UPR plays a role in apoptosis of lesional macrophages43 and regulation of inflammatory cytokines in aortic ECs.6 Our current findings raise the possibility that UPR signaling can also regulate angiogenesis characteristic of atherosclerosis. It is possible that the ATF4 mechanism can supplement hypoxia-induced HIF-1 cascade, thus amplifying formation of neovessels in atheroma. In support of this possibility, we show here that OxPL-induced UPR regulates VEGF independently of HIF-1. In addition to homocysteine, cholesterol, oxidative stress, and OxPLs,26,37,43 also very low oxygen levels were shown to up-regulate ATF4.56,57 These data suggest that ATF4 might serve as a point of convergence of different stress mechanisms coupling them to the angiogenic switch.
Activation of UPR has been characterized as a consequence but also a pathogenic mechanism in a variety of disease states, including tumors,41,58–60 suggesting that UPR may have broader implications in different types of pathologic angiogenesis.
In this work, we describe for the first time structural characteristics of OxPLs required for activation of UPR. In particular, the role of polar head groups and of sn-2 residues was analyzed. The structure of polar head group had minimal influence on UPR activation, suggesting that oxidation of any phospholipid class generates active species. In contrast, structural requirements for the sn-2 residue were more stringent. We found that the presence of oxidized but not native sn-2 residue was a prerequisite for induction of UPR genes because both unoxidized or lysophospholipids were characterized by a dramatically lower activity. Unesterified oxidized fatty acids mimicked effects of OxPLs on UPR induction, again characterizing oxidized sn-2 residue as the major determinant responsible for induction of UPR genes. The pathologic significance of the latter finding, however, requires testing whether free oxidized fatty acids accumulate in atherosclerotic vessels in concentrations comparable with OxPLs, known to reach the levels well above those inducing UPR in our experiments (total biologically active OxPLs were detected in atheroma locally at concentrations of hundreds of μmol/L4). We found that OxPLs containing variable fragmented or full-length oxidized residues were able to induce UPR genes, suggesting that phospholipids containing polyunsaturated fatty acids differing in chain length, as well as the number and location of double bonds, all are expected to form active species on oxidation. Last but not least, we ob-served very close parallels between structure-function relationships in respect to UPR and VEGF induction, thus again arguing for the role of UPR as a mechanism of VEGF up-regulation by OxPLs.
An important question relates to the mechanisms of cellular recognition of OxPLs leading to UPR initiation. As discussed in “Results” and supplemental materials, we rule out the role of several receptors previously implicated in biologic effects of OxPLs, including PAF-, lysophospholipid-, and EP2 prostaglandin receptors, PPARs, or TLR4. Our results rather point to the role of membrane effects, a paradigm opened previously by studies demonstrating activation of UPR by cholesterol and sphingolipids.38 However, we also cannot rule out the involvement of an unidentified pattern-recognition receptor with broad specificity for a wide range of oxygenated lipid molecules. Chaperones recognize short peptides typically found inside the hydrophobic interior of properly folded proteins but abnormally exposed on the surface of unfolded proteins.61,62 Therefore, it is possible that chaperones can bind hydrophobic phospholipid molecules as well. A candidate chaperone is GRP78/BIP known to recognize misfolded proteins inside the ER but also implicated as a signaling receptor located on the cell surface.63
In conclusion, we found that OxPLs stimulate expression of VEGF via activation of the ATF4 branch of UPR. Analysis of lipid specificity demonstrated that oxidized sn-2 residues play a key role in the activation of UPR/ATF4-dependent transcription and up-regulation of VEGF. Our findings further emphasize that UPR activated by atherogenic lipids, such as cholesterol or OxPLs, is an important signaling mechanism in atherosclerosis and a potential target for therapeutic interventions aiming at prevention of plaque growth as well as intra-plaque apoptosis and angiogenesis, both recognized as key determinants of plaque stability.
Supplementary Material
Acknowledgments
The authors thank Mario Hilpert for technical assistance.
This work was supported by Fonds zur Förderung wissenschaftlicher Forschung (S9407-B11, P18232-B11; V.N.B.), Jubiläumsfond der Österreichischen Nationalbank (AP12532ONB; O.V.O.), and the National Institutes of Health (grant HL30568; A.J.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: O.V.O., T.A., P.S.G., A.J.L., and V.N.B. participated in the design of the experiments; O.V.O., T.A., A.L., E.v.S., and P.S.G. performed research; O.V.O., T.A., P.S.G., A.J.L., and V.N.B. controlled and analyzed data; V.N.B. and B.R.B. drafted the manuscript; all authors checked the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Valery N. Bochkov, Department of Vascular Biology and Thrombosis Research, Medical University of Vienna, Schwarzspanierstrasse 17, A-1090 Vienna, Austria; e-mail: valery.bochkov@meduniwien.ac.at.
References
- 1.Hoff HF, O'Neil J, Wu Z, Hoppe G, Salomon RL. Phospholipid hydroxyalkenals: biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2003;23:275–282. doi: 10.1161/01.atv.0000051407.42536.73. [DOI] [PubMed] [Google Scholar]
- 2.Podrez EA, Poliakov E, Shen Z, et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 3.Subbanagounder G, Leitinger N, Schwenke DC, et al. Determinants of bioactivity of oxidized phospholipids: specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000;20:2248–2254. doi: 10.1161/01.atv.20.10.2248. [DOI] [PubMed] [Google Scholar]
- 4.Watson AD, Leitinger N, Navab M, et al. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 5.Bochkov VN. Inflammatory profile of oxidized phospholipids. Thromb Haemost. 2007;97:348–354. [PubMed] [Google Scholar]
- 6.Gargalovic PS, Gharavi NM, Clark MJ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 7.Bochkov VN, Kadl A, Huber J, et al. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 8.Kronke G, Bochkov VN, Huber J, et al. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 9.Birukov KG, Bochkov VN, Birukova AA, et al. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 10.Nonas S, Miller I, Kawkitinarong K, et al. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury. Am J Respir Crit Care Med. 2006;173:1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies SS, Pontsler AV, Marathe GK, et al. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma ligands and agonists. J Biol Chem. 2001;276:16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 12.Li R, Mouillesseaux KP, Montoya D, et al. Identification of prostaglandin E2 receptor subtype 2 as a receptor activated by OxPAPC. Circ Res. 2006;98:642–650. doi: 10.1161/01.RES.0000207394.39249.fc. [DOI] [PubMed] [Google Scholar]
- 13.Marathe GK, Prescott SM, Zimmerman GA, McIntyre TM. Oxidized LDL contains inflammatory PAF-like phospholipids. Trends Cardiovasc Med. 2001;11:139–142. doi: 10.1016/s1050-1738(01)00100-1. [DOI] [PubMed] [Google Scholar]
- 14.Walton KA, Hsieh X, Gharavi N, et al. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8: a role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 15.Birukov KG. Oxidized lipids: the two faces of vascular inflammation. Curr Atheroscler Rep. 2006;8:223–231. doi: 10.1007/s11883-006-0077-x. [DOI] [PubMed] [Google Scholar]
- 16.Leitinger N. Oxidized phospholipids as triggers of inflammation in atherosclerosis. Mol Nutr Food Res. 2005;49:1063–1071. doi: 10.1002/mnfr.200500086. [DOI] [PubMed] [Google Scholar]
- 17.Bochkov VN, Philippova M, Oskolkova O, et al. Oxidized phospholipids stimulate angiogenesis via autocrine mechanisms, implicating a novel role for lipid oxidation in the evolution of atherosclerotic lesions. Circ Res. 2006;99:900–908. doi: 10.1161/01.RES.0000245485.04489.ee. [DOI] [PubMed] [Google Scholar]
- 18.Gimbrone MA, Jr, Cotran RS, Folkman J. Human vascular endothelial cells in culture: growth and DNA synthesis. J Cell Biol. 1974;60:673–684. doi: 10.1083/jcb.60.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milne GL, Seal JR, Havrilla CM, Wijtmans M, Porter NA. Identification and analysis of products formed from phospholipids in the free radical oxidation of human low density lipoproteins. J Lipid Res. 2005;46:307–319. doi: 10.1194/jlr.M400311-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Gu X, Sun M, Gugiu B, et al. Oxidatively truncated docosahexaenoate phospholipids: total synthesis, generation, and peptide adduction chemistry. J Org Chem. 2003;68:3749–3761. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- 21.Mihaljevic B, Katusin-Razem B, Razem D. The reevaluation of the ferric thiocyanate assay for lipid hydroperoxides with special considerations of the mechanistic aspects of the response. Free Radic Biol Med. 1996;21:53–63. doi: 10.1016/0891-5849(95)02224-4. [DOI] [PubMed] [Google Scholar]
- 22.Broekhuyse RM. Phospholipids in tissues of the eye: I. Isolation, characterization and quantitative analysis by two-dimensional thin-layer chromatography of diacyl and vinyl-ether phospholipids. Biochim Biophys Acta. 1968;152:307–315. doi: 10.1016/0005-2760(68)90038-6. [DOI] [PubMed] [Google Scholar]
- 23.Ema M, Taya S, Yokotani N, et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bochkov VN, Mechtcheriakova D, Lucerna M, et al. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca(++)/NFAT. Blood. 2002;99:199–206. doi: 10.1182/blood.v99.1.199. [DOI] [PubMed] [Google Scholar]
- 25.Kadl A, Huber J, Gruber F, et al. Analysis of inflammatory gene induction by oxidized phospholipids in vivo by quantitative real-time RT-PCR in comparison with effects of LPS. Vascul Pharmacol. 2002;38:219–227. doi: 10.1016/s1537-1891(02)00172-6. [DOI] [PubMed] [Google Scholar]
- 26.Roybal CN, Hunsaker LA, Barbash O, Vander Jagt DL, Abcouwer SF. The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4-dependent mechanism. J Biol Chem. 2005;280:20331–20339. doi: 10.1074/jbc.M411275200. [DOI] [PubMed] [Google Scholar]
- 27.Siu F, Bain PJ, LeBlanc-Chaffin R, Chen H, Kilberg MS. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J Biol Chem. 2002;277:24120–24127. doi: 10.1074/jbc.M201959200. [DOI] [PubMed] [Google Scholar]
- 28.Yoo PS, Mulkeen AL, Cha CH. Post-transcriptional regulation of vascular endothelial growth factor: implications for tumor angiogenesis. World J Gastroenterol. 2006;12:4937–4942. doi: 10.3748/wjg.v12.i31.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 30.Moulton KS. Plaque angiogenesis and atherosclerosis. Curr Atheroscler Rep. 2001;3:225–233. doi: 10.1007/s11883-001-0065-0. [DOI] [PubMed] [Google Scholar]
- 31.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 32.Paul SA, Simons JW, Mabjeesh NJ. HIF at the crossroads between ischemia and carcinogenesis. J Cell Physiol. 2004;200:20–30. doi: 10.1002/jcp.10479. [DOI] [PubMed] [Google Scholar]
- 33.Shatrov VA, Sumbayev VV, Zhou J, Brune B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101:4847–4849. doi: 10.1182/blood-2002-09-2711. [DOI] [PubMed] [Google Scholar]
- 34.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 35.Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]
- 36.Tintut Y, Parhami F, Tsingotjidou A, et al. 8-Isoprostaglandin E2 enhances receptor-activated NFkappa B ligand (RANKL)-dependent osteoclastic potential of marrow hematopoietic precursors via the cAMP pathway. J Biol Chem. 2002;277:14221–14226. doi: 10.1074/jbc.M111551200. [DOI] [PubMed] [Google Scholar]
- 37.Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Oyadomari S. Lipid phase perturbations and the unfolded protein response. Dev Cell. 2004;7:287–288. doi: 10.1016/j.devcel.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Tessitore A, del P Martin M, Sano R, et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 41.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Zhang C, Cai Y, Adachi MT, et al. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867–35874. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- 44.Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1:109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chevalier M, Rhee H, Elguindi EC, Blond SY. Interaction of murine BiP/GRP78 with the DnaJ homologue MTJ1. J Biol Chem. 2000;275:19620–19627. doi: 10.1074/jbc.M001333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroczynska B, Evangelista CM, Samant SS, Elguindi EC, Blond SY. The SANT2 domain of the murine tumor cell DnaJ-like protein 1 human homologue interacts with alpha1-antichymotrypsin and kinetically interferes with its serpin inhibitory activity. J Biol Chem. 2004;279:11432–11443. doi: 10.1074/jbc.M310903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang HY, Wek SA, McGrath BC, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 51.Lucerna M, Zernecke A, de Nooijer R, et al. Vascular endothelial growth factor-A induces plaque expansion in ApoE knock-out mice by promoting de novo leukocyte recruitment. Blood. 2007;109:122–129. doi: 10.1182/blood-2006-07-031773. [DOI] [PubMed] [Google Scholar]
- 52.Moulton KS. Angiogenesis in atherosclerosis: gathering evidence beyond speculation. Curr Opin Lipidol. 2006;17:548–555. doi: 10.1097/01.mol.0000245261.71129.f0. [DOI] [PubMed] [Google Scholar]
- 53.Nawa T, Nawa MT, Adachi MT, et al. Expression of transcriptional repressor ATF3/LRF1 in human atherosclerosis: colocalization and possible involvement in cell death of vascular endothelial cells. Atherosclerosis. 2002;161:281–291. doi: 10.1016/s0021-9150(01)00639-6. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 55.Ramos MA, Kuzuya M, Esaki T, et al. Induction of macrophage VEGF in response to oxidized LDL and VEGF accumulation in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1998;18:1188–1196. doi: 10.1161/01.atv.18.7.1188. [DOI] [PubMed] [Google Scholar]
- 56.Ameri K, Lewis CE, Raida M, et al. Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood. 2004;103:1876–1882. doi: 10.1182/blood-2003-06-1859. [DOI] [PubMed] [Google Scholar]
- 57.Blais JD, Filipenko V, Bi M, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–614. doi: 10.1016/s0168-8278(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 60.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 61.Blond-Elguindi S, Cwirla SE, Dower WJ, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 62.Flynn GC, Pohl J, Flocco MT, Rothman JE. Peptide-binding specificity of the molecular chaperone BiP. Nature. 1991;353:726–730. doi: 10.1038/353726a0. [DOI] [PubMed] [Google Scholar]
- 63.Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006;281:13694–13707. doi: 10.1074/jbc.M511694200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}