

Abstract
Phospholipids when dispersed in excess water generally form vesicular membrane structures. Cryo-transmission and freeze-fracture electron microscopy are combined here with calorimetry and viscometry to demonstrate the reversible conversion of phosphatidylglycerol aqueous vesicle suspensions to a three-dimensional structure that consists of extended bilayer networks. Thermodynamic analysis indicates that the structural transitions arise from two effects: (i) the enhanced membrane elasticity accompanying the lipid state fluctuations on chain melting and (ii) solvent-associated interactions (including electrostatics) that favor a change in membrane curvature. The material properties of the hydrogels and their reversible formation offer the possibility of future applications, for example in drug delivery, the design of structural switches, or for understanding vesicle fusion or fission processes.
Lipids may undergo a cooperative melting reaction linked to the loss in conformational order of the lipid chains (1). This transition is accompanied by a large change of the internal energy (or enthalpy) that can be studied by calorimetry. It has long been known that the melting profiles are influenced by secondary components, e.g., proteins (2), cholesterol, or anesthetics (3, 4). Phase diagrams of lipid mixtures usually are constructed as a function of the chemical potential of the components and the temperature (5). Another kind of possible transition involves changes in vesicular shape. These transitions have been studied in some detail by theoretical means, resulting in phase diagrams for vesicle structure as a function of the elastic constants (6, 7). Changes in these constants can explain theoretically the wealth of shapes of erythrocytes (8). There are therefore two kinds of phase diagrams found for lipid systems: one describing the calorimetric features of chain melting, mainly dependent on short-range cooperative events within the membrane plane, and the other describing transitions in shape and long-range order influenced by changes in the elastic constants.
Although seemingly different features of a lipid system, the elastic constants and the heat capacity of the lipid membrane are related because they are coupled to thermodynamic fluctuations in the membrane properties (9). Maximum elasticity is achieved in the regime of ordered (gel)-fluid phase coexistence (10), which implies that close to the heat capacity maximum the elastic constants change markedly and as a consequence the membranes become more flexible. From this it is expected that changes in structural equilibria may be found close to the chain-melting transition.
We demonstrate here thermodynamically how the structural changes couple to the chain melting. Large viscosity changes of dimyristoyl phosphatidylglycerol (DMPG) dispersions close to the calorimetric melting transition have been reported that are accompanied by marked changes in heat capacity, light scattering, and electrical conductance (11, 12). The changes in viscosity suggest a change in the long-range interactions of the lipid membranes and large-scale geometrical changes. We show here definitively, both by cryo- and freeze-fracture electron microscopy, that the viscous state of DMPG dispersions consists of an extended three-dimensional bilayer network. Thus, a high-viscosity hydrogel is formed in the phase coexistence region that converts reversibly to low-viscosity vesicular suspensions at the two extremes of the broadened chain-melting region. The extended networks bear a morphological resemblance to the lattice networks of tubular myelin that are formed by lung surfactant in the alveolar fluid. Lung surfactant is uniquely rich in dipalmitoyl phosphatidylcholine and phosphatidylglycerol, whose chain melting occurs close to physiological temperatures (13–15). Both the reversibility of the material properties demonstrated here, and the possible connection with the pulmonary surfactant cycle offer possibilities for future exploitation, e.g., in liposomal drug delivery and the design of synthetic lung surfactant substitutes. The morphologically observed changes in membrane curvature provide the key to interpreting the coupling between structure, elasticity, and thermodynamics of chain melting.
Materials and Methods
Anionic phospholipids in their sodium form were purchased from Avanti Polar Lipids and used without further purification. Lipids were dispersed in a 2 mM Hepes (Sigma) 1 mM EDTA (Fluka) buffer at pH 7.5. The dispersions were incubated at 45°–60°C for several minutes and vortexed after incubation. Extended lipid phases formed spontaneously. The ionic strength of the buffer generally is given in concentrations of Na+-ions including counterions of the buffer (2 mM Hepes, 1 mM EDTA, pH 7.5). Some experiments were performed in distilled water to keep the ionic strength low. The pH was checked in those experiments.
Calorimetry.
Heat capacity profiles were recorded on a MC2 calorimeter of Microcal (Northampton, MA) (Fig. 1) or on a four-cell calorimeter of Calorimetric Science (Provo, Utah) (see Fig. 3), by using a scan rate of 5°/h.
Figure 1.

Schematic summary of the chain of events deduced from calorimetry, viscosity measurements, and electron microscopy (see Figs. 2 and 3). Structural changes represent changes in the three-dimensional membrane arrangement. These changes correlate with viscosity increases and distinct heat capacity maxima. Viscosities are given relative to those of water at the same temperature.
Figure 3.

(Left) Heat capacity traces of a 10 mM DMPG dispersion under various ionic strength conditions (in a 2 mM Hepes, 1 mM EDTA, pH 7.5 buffer; the top trace was measured in distilled water). At low ionic strength the Cp traces show a complex behavior as in Fig. 1. With increasing NaCl concentration the chain of events occurs over a narrower temperature interval. At 500 mM Na+ one single, highly cooperative heat capacity peak is found. Below the main transition the pretransition can be found (indicated by an arrow). This low enthalpy transition is not considered here in detail. It is, however, also linked to minor structural changes (formation of ripples on the surface, visible on the membrane surface in Fig. 2B Left. (Right) Heat capacity traces of 10 mM dispersions of phosphatidylglycerols with various chain lengths (in distilled water, pH ≈7.5). Di(tridecanoyl) PG (C13 chains) and dimyristoyl PG (C14 chains) show a complex behavior as in Fig. 1. With increasing chain length the chain of events occurs over a narrower temperature interval. Dipalmitoyl PG (C16 chains) displays only one very cooperative heat capacity peak.
Viscosity Measurements.
Viscosities were measured with a Contraves Low Shear 30 viscometer (Stuttgart, Germany) by using a concentric cylinder pendulum (set 1 cup/bob combination). The viscosity of the lipid dispersion is given relative to the viscosity of water at the same temperature.
Electron Microscopy.
The sample dispersions were rapidly frozen in liquid ethane or propane with an estimated freezing rate above 104 K⋅s−1. Freeze fracture electron microscopy was performed by using a Philips 301 electron microscope. After fracturing the support sandwich, the surface was shadowed unidirectionally (45° angle) by Ta-W metal vapor and coated with a 200-Å carbon film. Five percent glycerol was added to the buffer to prevent ice crystal formation. For details see ref. 16. Cryo-transmission electron microscopy (cryo-TEM) was performed by using a Philips CM12 electron microscope. Details of the cryo-TEM procedure are given in ref. 17.
Experimental Findings
Fig. 1 summarizes some of our experimental findings. Aqueous dispersions of the charged lipid DMPG at low ionic strength display an extended and complex melting behavior, with three maxima in the melting regime (Fig. 1, lower trace). The heat of transition integrated over the entire region (≈22 kJ/mol) corresponds to that reported for the chain-melting reaction. The two outer maxima in the heat capacity coincide with extremely marked changes in the viscosity of the dispersion (Fig. 1, upper trace). Depending on temperature and ionic strength, the viscosity increase may be up to 80-fold (data not shown). This striking change in material properties of the lipid suspensions is related to a structural transition from a vesicular state below the transition to a long-range bilayer network in the transition region. Above the range of calorimetric events, vesicles, but of different morphology, again prevail (see Fig. 2, discussed below). Throughout the transition region, the lipid membranes assume an entirely different topology, forming an extended phase with different mean curvature (Fig. 1 Top). The structural changes are evident in electron microscopy. We have performed freeze fracture (16) and cryo-TEM (17) of dispersions by using buffer conditions similar to those in Fig. 1. The two techniques are complementary because cryo-TEM gives a view through the sample, whereas freeze-fracture electron microscopy reveals individual membrane surfaces. At temperatures below the melting transition, large unilamellar vesicles with mean diameters around 500–1,000 nm are found (Fig. 2A). They are not spherical but are facetted and polyhedral in shape. Above the melting regime, the dispersion becomes again vesicular. However, the size distribution is centered around a lower diameter (50–200 nm) and the vesicles are now largely spherical (Fig. 2C). The different vesicular geometry is a consequence of the different bending elasticities for the ordered (gel) and the fluid phases (18, 19). The lipid phase in the melting regime is not vesicular and no localized bounded structures can be seen (Fig. 2B). Large stacks of membranes with large interlamellar spacings extend throughout the whole sample, in a network-like manner. From phosphorus NMR it is known that the extended phase is lamellar (11). Although any long-range symmetry of the extended structure still remains to be resolved, it clearly possesses different connectivity, symmetry, topology, and mean surface curvature from the vesicular phases below and above the melting regime.
Figure 2.

(Left) Freeze fracture electron micrographs of a 45 mM DMPG dispersion. (A) At 9°C, displaying large vesicles (representative features of different fields are given). The membrane sheets are overlaid by short periodic ripples. (B) At 24.5°C, displaying membrane sheets, arranged randomly toward the fractured surface and continuous membrane segments (representative features of different fields are given). The membrane sheets are overlaid by large periodic ripples. In some segments these ripples can be seen in a cross section. (C) At 50°C, displaying perfectly spherical vesicles of various sizes (representative features of different fields are given). The vesicles on average are significantly smaller than in A. (Right) Cryo-TEM micrographs of a 15 mM DMPG dispersion. (A) At 15°C, displaying large unilamellar vesicles (representative features of different fields are given) that are not round in shape. (B) At 25°C, displaying randomly curved (Right) or regularly spaced (Left) continuous membranes (representative features of different fields are given). (C) At 45°C, displaying unilamellar vesicles of various sizes (representative features of different fields are given). Some vesicles are situated within larger vesicles.
It should be noted that these network structures, which are obtained only in the chain-melting region, are quite distinct from those reported by Kodama and coworkers (20, 21). The latter tubular structures are obtained after prolonged incubation at 5°C (up to 30 days), resulting in formation of the crystalline or sub-gel phase of DMPG that has a chain-melting transition that occurs at much higher temperature and is not reversible on cooling.
The complex phase behavior that is associated with the network structures depends strongly on environmental conditions. Increasing the ionic strength lowers the width of the temperature range over which the viscous phase exists (Fig. 3 Left). The viscosity changes occur in the same regime as the excess heat capacity events of the main transition. Data from our laboratory reveal viscosity changes in the melting regime with boundaries coinciding with the calorimetric events. Their magnitude depends largely on the ionic strength (data not shown). Light scattering data display a similar behavior, with a pronounced decrease in scattering intensity in the melting regime (22). A reduction in temperature range over which the extended phase is stable results from lowering the solvent activity. Increase in lipid concentration (11, 23) or addition of osmotically active polymers (24) (upon addition of 30% polyethylene glycol at low ionic strength the heat capacity profile displays only a single peak) narrows the melting regime. Because variation in buffer conditions controls the stability range of the extended phase, it can be concluded that the intermediate state interacts differently with the solvent than do the vesicular states below and above the transition. The free energy difference of this interaction, ΔGsolv, is shown below to explain the coupling between chain melting and transitions in vesicular morphology. It should be stressed that this additional interaction, and not just the changes in membrane elasticity on chain melting, is essential to explain the morphological changes observed. Of course, ΔGsolv includes primarily the electrostatic interactions, which themselves strongly depend on the solvent (see e.g., ref. 1), and also contribute to the overall osmotic equilibrium.
An intrinsic factor influencing the transition behavior is the lipid chain length (Fig. 3 Right). The shorter the chains, the broader is the melting regime. Dipalmitoyl phosphatidylglycerol with C16 acyl chains displays only one melting peak at low ionic strength, whereas the homologue with C13 chains [di(tridecanoyl) phosphatidylglycerol] possesses a melting range extending over more than 30°C. This dependence on membrane composition therefore reflects both the heat content and the cooperativity of the chain-melting event, both of which differ with chain length.
Finally, we note that these findings are not restricted either to phosphatidylglycerols or solely to anionic phospholipids, although the effects are generally most pronounced in these cases. Dimyristoyl phosphatidylserine and ditetradecyl phosphatidylglycerol (ether linked chains) at low concentrations and low ionic strength display two calorimetric peaks (data not shown). Complexes of the angiotension receptor antagonist Losartan with vesicles of the zwitterionic phospholipid dimyristoyl phosphatidylcholine display calorimetric behavior and formation of a high-viscosity phase similar to that found here for DMPG (25). This behavior may in part be determined by the charge on the drug molecule. Recently, similar, although more attenuated, calorimetric and viscosity changes have been found with extruded unilamellar vesicles of dimyristoyl phosphatidylcholine alone. (Note that the DMPG vesicles in Fig. 2 are also unilamellar.) Extruded 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) vesicles display two calorimetric peaks in the melting regime which extends over ca. 3° and is accompanied by a 10% viscosity change. The corresponding heat capacity trace is given in ref. 10. This result indicates that extruded vesicles of DMPC possess the potential for a morphological transition, but lack the strong osmotic driving force that is inherent to charged lipid vesicles. Thus, the formation of high-viscosity extended three-dimensional lipid phases emerges potentially as a rather general feature arising from the coupling of chain melting with a solvent-related driving force that modulates membrane curvature. Variation in temperature, ionic strength, chain composition, and protein binding can serve as triggers for these structural changes.
Discussion
Why Do Structural Changes Occur Close to the Melting Transition?
Membrane area compressibilities (19, 26) and correspondingly the bending elasticity (27, 28) increase greatly in the chain-melting regime. This phenomenon, established experimentally for unilamellar phosphatidylcholine vesicles, is likely to be general for all lipids, because it arises from the energetically facile interconversion of the coexisting lipid states that enhances the fluctuations in membrane area at the melting point. The compressibility is defined as the change of membrane area with respect to lateral pressure changes. The bending elasticity similarly reflects the change in the free energy of a membrane segment with respect to the membrane curvature. The morphological results (Fig. 2) indicate that it is the curvature elasticity that is of primary relevance, because the transitions between the different structural states involve pronounced changes in membrane curvature (curved vesicles are transformed into bilayer networks with low curvature). For simplicity, we consider an equilibrium between two lipid membrane segments with different mean curvature, one planar and the other spherical, controlled by a free energy difference ΔG(T) (see Fig. 4).
Figure 4.

Schematic representation of the temperature-dependent equilibrium between a flat and a curved membrane segment.
This free energy difference has two contributions that display very different temperature dependence. Bending of the membrane segment from the planar to the curved geometry costs a bending free energy contribution, ΔGelast, which is a strong function of temperature when chain melting takes place. In the absence of other interactions, the planar state is the more stable for a symmetric membrane. The free energy penalty of bending, however, is much less in the chain-melting regime (10) as demonstrated experimentally to be generally the case by comparison of lipids with different chain-melting temperatures (27). The experimental findings reported above (Fig. 3 Left) indicate that at low ionic strength and low lipid concentrations there are additional interactions that tend to change the membrane curvature. These solvent-dependent interactions include electrostatics and are designated globally by ΔGsolv, which has a sign such as to shift the equilibrium over to the right in the model situation given above. Only when ΔGelast is drastically reduced in the chain-melting region is this “osmotic driving force,” ΔGsolv, sufficiently strong to give rise to a structural change. With these simple principles, it is possible to explain the complex calorimetric behavior observed during chain melting. Purely for illustration, a symmetrical membrane for which the curved membrane has higher free energy is assumed, although this may not necessarily correspond to the experimental situation depicted in Fig. 1. However, the general thermodynamic arguments do not depend for their validity on the detailed nature of the change in mean curvature nor of the osmotic driving force.
Modeling the Transition Behavior Between Two Membrane Segments of Different Curvature.
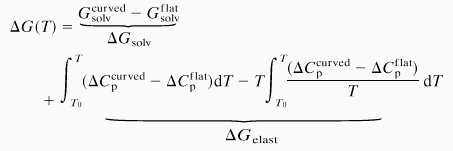
Experimentally it has been found that the heat capacity profiles of the lipid melting process are broadened for curved membranes (10, 29). This broadening reflects that the work necessary to bend the membrane is temperature dependent (Fig. 5 a and c, Lower). The heat capacity profiles shown are actually calculated from a two-dimensional Ising model (30) involving two individual lipid states (ordered and fluid), with the melting process described by Monte-Carlo simulations (2, 31). Curvature is introduced by constraining the relative populations of the two lipid states (which have different molecular areas) to be different in the two monolayer halves of the bilayer. The actual area asymmetry assumed corresponds to a mean radius of membrane curvature of approximately 60 nm. Because the largest contribution to the elasticity in the melting regime stems from fluctuations in lipid state (10), we neglect other contributions and assume that bending is synonymous with an asymmetry in the number of fluid lipids in both monolayers. From these heat capacity profiles, we have calculated the temperature dependence of the membrane free energy for different values of the “osmotic driving force,” ΔGsolv, by using the model introduced in the previous section. For the flat and curved membranes, the free energies and their difference are given respectively by:
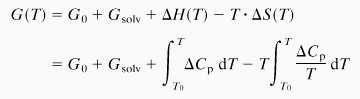 |
1 |
 |
2 |
where ΔGsolv = Gsolvcurved − Gsolvflat is assumed for simplicity to have little temperature dependence and to display no discontinuity on chain melting. The last two terms in Eq. 2 reflect the bending free energy difference, ΔGelast, of a curved and a flat membrane. Results for G(T) for flat and curved membranes are given by the blue and red curves, respectively, in the upper parts of Fig. 5. Solid lines indicate the lower free energy state. The nonsolvent contributions to the free energy are calculated directly from the heat capacity profiles in Fig. 5 a and c by using Eq. 1 for flat and curved membranes, respectively. The free energy profiles given in Fig. 5 differ only in the values of ΔGsolv, which affects the relative stability of the planar and curved membranes. In Fig. 5b, the value of ΔGsolv is chosen such that the two free energy profiles intersect and structural transitions take place at the points of intersection, whose position depends in detail on the value of ΔGsolv. The resulting value of ΔG(T) is given in the center part of Fig. 5b. By differentiating (2) with respect to temperature, the heat capacity profile given in Fig. 5b is obtained from the free energy difference. This calorimetric profile contains the three characteristic peaks observed experimentally for DMPG (Fig. 1). Of course, there are differences in detail from the idealized example. In particular, the experimental profiles are not symmetric because vesicular morphologies in the low and high temperature regions differ considerably (Fig. 2). Nevertheless, the general principles of the modulation of the chain melting by coupling to structural transitions induced by changes in the membrane elasticity are established by this model calculation. A broadened chain-melting regime and transitions to structures of different mean curvature, in the present case to extended bilayer networks, are to be expected whenever external conditions are such as to augment the decrease in curvature energy inherent in the coexistence region of different lipid states. This thermodynamic reasoning is quite general and does not depend on the details of the Monte-Carlo simulations that are used for this illustration. One may equally well use experimental heat capacity profiles for differently curved systems (10, 29). Nor does it depend on specific characteristics of the morphological change, or the origin of the osmotic driving force.
Figure 5.

Calculation of the free energy and the heat capacities for membranes that are constrained to be flat or curved with a radius of curvature of r = 60 nm (from a Monte Carlo simulation, using Eqs. 1 and 2). The free energy of the curved state is given in red, that of the flat state in blue. The lower free energy state is shown as solid lines, the higher energy states as dotted lines. The interaction with the solvent is taken to be different for the two states. This difference, ΔGsolv, differs between a–c, leading to a relative shift in the vertical direction between the free energy profiles of the flat and the curved states. (a) Flat membrane with one narrow heat capacity peak. (b) Coexistence of flat and curved membranes, leading to a structural transition at the intersection points of the free energy curves. (c) Curved membrane with a broad heat capacity peak.
Conclusions
A membrane structure that consists of three-dimensional extended bilayer networks is produced reversibly during chain melting of lipid dispersions, when solvent conditions are such as to favor curvature. The variety of conditions under which network formation can occur is quite diverse, and theoretical considerations suggest that structural changes induced by chain melting are a general phenomenon. Undoubtedly, further conditions will be found that induce structural changes or network formation. The theoretical analysis given of the heat capacity profiles suggests that calorimetry will prove to be a most valuable tool in the continuing search for further comparable systems. The special material properties of these hydrogel structures, their high viscosity, and judging from the elasticity analysis, their high intrinsic flexibility, coupled with the reversibility of their formation, warrants consideration for innovative applications. The inherent broadening of the chain-melting regime that accompanies their formation further enhances their potential. These materials are “intelligent” materials whose macroscopic properties can be switched as a function of ionic strength, temperature, or drug binding. Similar processes may be important in fusion and fission of lipid vesicles and possibly even biological membranes. The analogy with pulmonary surfactant and possible application of these principles in surfactant supplement formulations for treatment of infantile respiratory syndrome remains to be explored. Other intriguing features such as the impact on liposomal drug delivery systems (release of encapsulated drugs may be triggered by network formation) offer further opportunity for exploitation of these unique structures. The exact nature of the osmotic driving forces favoring morphological transitions, and the diversity of their possible origin, also are areas that still remain to be explored.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG).
Abbreviations
- cryo-TEM
cryo-transmission electron microscopy
- DMPG
dimyristoyl phosphatidylglycerol
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cevc G, Marsh D. Phospholipid Bilayers: Physical Principles and Models. New York: Wiley; 1987. [Google Scholar]
- 2.Heimburg T, Biltonen R L. Biophys J. 1996;70:84–96. doi: 10.1016/S0006-3495(96)79551-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMullen T P W, McElhaney R N. Biochim Biophys Acta. 1995;1234:90–98. doi: 10.1016/0005-2736(94)00266-r. [DOI] [PubMed] [Google Scholar]
- 4.Mouritsen O G, Jørgensen K. Mol Membr Biol. 1995;12:15–20. doi: 10.3109/09687689509038490. [DOI] [PubMed] [Google Scholar]
- 5.Lee A G. Biochim Biophys Acta. 1977;472:285–344. doi: 10.1016/0304-4157(77)90001-6. [DOI] [PubMed] [Google Scholar]
- 6.Helfrich W. J Phys Condens Matter. 1994;6:A79–A92. [Google Scholar]
- 7.Seifert U, Lipowsky R. In: Structure and Dynamics of Membranes: From Cells to Vesicles. Lipowsky R, Sackmann E, editors. Amsterdam: Elsevier; 1995. pp. 403–463. [Google Scholar]
- 8.Sackmann, E. in Structure and Dynamics of Membranes: From Cells to Vesicles, eds. Lipowsky, R. & Sackmann, E. (Elsevier, Amsterdam), pp. 213–304.
- 9.Halstenberg S, Heimburg T, Hianik T, Kaatze U, Krivanek R. Biophys J. 1998;75:264–271. doi: 10.1016/S0006-3495(98)77513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heimburg T. Biochim Biophys Acta. 1998;1415:147–162. doi: 10.1016/s0005-2736(98)00189-8. [DOI] [PubMed] [Google Scholar]
- 11.Heimburg T, Biltonen R L. Biochemistry. 1994;33:9477–9488. doi: 10.1021/bi00198a013. [DOI] [PubMed] [Google Scholar]
- 12.Kodama M, Miyata T. Thermochim Acta. 1995;267:365–372. [Google Scholar]
- 13.Sanders R L, Hassett R J, Vatter A E. Anat Rec. 1980;198:485–501. doi: 10.1002/ar.1091980310. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki Y, Fujita Y, Kogishi K. Am Rev Respir Dis. 1989;140:75–81. doi: 10.1164/ajrccm/140.1.75. [DOI] [PubMed] [Google Scholar]
- 15.Williams M C. In: Pulmonary Surfactant: From Molecular Biology to Clinical Practice. Robertson B, Van Golde L M G, Batenberg J J, editors. Amsterdam: Elsevier; 1992. pp. 87–107. [Google Scholar]
- 16.Strey R, Jahn W, Porte G, Bassera P. Langmuir. 1990;6:1635–1639. [Google Scholar]
- 17.Talmon Y. Ber Bunsenges Phys Chem. 1996;100:364–372. [Google Scholar]
- 18.Evans E A. Biophys J. 1974;14:923–931. doi: 10.1016/S0006-3495(74)85959-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans E A, Kwok R. Biochemistry. 1982;21:4874–4879. doi: 10.1021/bi00263a007. [DOI] [PubMed] [Google Scholar]
- 20.Kodama M, Miyata T, Yokoyama T. Biochim Biophys Acta. 1993;1168:243–248. [PubMed] [Google Scholar]
- 21.Kodama M, Aoki H, Miyata T. Biophys Chem. 1999;79:205–217. doi: 10.1016/s0301-4622(99)00053-8. [DOI] [PubMed] [Google Scholar]
- 22.Riske K A, Politi M J, Reed W F, Lamy-Freund M T. Chem Phys Lip. 1997;89:31–44. [Google Scholar]
- 23.Kodama M, Nakamura J, Miyata T, Aoki H. J Therm Anal. 1998;51:91–104. [Google Scholar]
- 24.Henze J. Diploma Thesis. Germany: University of Göttingen; 1994. [Google Scholar]
- 25.Theodoropoulou, E. & Marsh, D. (1999) Biochim. Biophys. Acta, in press. [DOI] [PubMed]
- 26.Needham D, Evans E. Biochemistry. 1988;27:8261–8269. doi: 10.1021/bi00421a041. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Puente L, Bivas I, Mitov M D, Méléard P. Europhys Lett. 1994;28:181–186. [Google Scholar]
- 28.Méléard P, Gerbeaud C, Pott T, Fernandez-Puente L, Bivas I, Mitov M D, Dufourcq J, Bothorel P. Biophys J. 1997;72:2616–2629. doi: 10.1016/S0006-3495(97)78905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brumm T, Jørgensen K, Mouritsen O G, Bayerl T M. Biophys J. 1996;70:1373–1379. doi: 10.1016/S0006-3495(96)79695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doniach S. J Chem Phys. 1978;68:4912–4916. [Google Scholar]
- 31.Sugar I P, Biltonen R L, Mitchard N. Methods Enzymol. 1994;240:569–593. doi: 10.1016/s0076-6879(94)40064-4. [DOI] [PubMed] [Google Scholar]