

Abstract
Adequate cochlear blood supply by the spiral modiolar artery (SMA) is critical for normal hearing. ACh may play a role in neuroregulation of the SMA but several key issues including its membrane action mechanisms remain poorly understood. Besides its well-known endothelium-dependent hyperpolarizing action, ACh can induce a depolarization in vascular cells. Using intracellular and whole-cell recording techniques on cells in guinea pig in vitro SMA, we studied the ionic mechanism underlying the ACh-depolarization and found that: 1) ACh induced a DAMP-sensitive depolarization when intermediate conductance KCa channels were blocked by charybdotoxin or nitrendipine. The ACh-depolarization was associated with a decrease in input resistance (Rinput) in high membrane potential (Vm) (~−40 mV) cells but with no change or an increase in Rinput in low Vm (~−75 mV) cells. ACh-depolarization was attenuated by background membrane depolarization from ~−70 mV in the majority of cells; 2) ACh-induced inward current in smooth muscle cells embedded in a SMA segment often showed a U-shaped I/V curve, the reversal potential of its two arms being near EK and 0 mV respectively; 3) ACh-depolarization was reduced by low Na+, zero K+ or 20 mM K+ bath solutions; 4) ACh-depolarization was inhibited by La3+ in all cells tested, by 4-AP and flufenamic acid in low Vm cells, but was not sensitive to Cd2+, Ni2+, nifedipine, niflumic acid, DIDS, IAA94, linopirdine or amiloride. We conclude that ACh-induced vascular depolarization was generated mainly by activation of a TRP-like non-selective cation channel and by inactivation of an inward rectifier K+ channel.
Keywords: membrane potential, intracellular recording, whole-cell recording, TRP channel, inward rectifier potassium channel, arteriole
1. Introduction
Auditory transduction is associated with heavy energy consumption (Thalmann et al., 1972) and is therefore particularly sensitive to any reduction in cochlear blood flow. Strong evidence shows that vascular disease or dysfunction is a major contributing factor to hearing loss (Hultcrantz, 1988; Nuttall, 1999; Schuknecht, 1982). The cochlear spiral modiolar artery (SMA) is of particular importance, because it supplies nearly all blood flow to the cochlea (Axelsson, 1988). A clog in the SMA will result in a cochlear stroke and sudden hearing loss. A spasm may result in cochlear dysfunction, causing symptoms such as vertigo and tinnitus (Baguley, 2002). Yet, current poor understanding of neurohumoral regulation of the SMA significantly limits our understanding of the cochlear vascular dysfunction that underlies numerous cochlear diseases.
We demonstrated (Jiang et al., 2005) that cholinergic fibers and varicosities form a sparse plexus in the adventitial layer of the SMA, and an M3 receptor antagonist 4-DAMP inhibits the evoked excitatory junction potential (EJP) in the vascular smooth muscle cells (VSMCs) that have a highly polarized membrane potential (~−75 mV) in the SMA. It is uncertain whether the classic neurotransmitter acetylcholine (ACh) plays a role in neuroregulation of the SMA. Identification of the ionic mechanism of ACh membrane actions would be an important step towards the understanding of the role.
ACh exerts opposite dual actions on the cochlear spiral modiolar artery (Jiang et al., 2001) and some other vascular tissues (Beny et al., 1988; Jaiswal et al., 1991). ACh hyperpolarizes and relaxes the SMA when its endothelial function is intact, whereas it causes a depolarization and contraction in functionally endothelium-denuded vessels (Jiang et al., 2007; Jiang et al., 2005). The ACh-induced hyperpolarization and relaxation have been attributed to the so-called “endothelium-derived hyperpolarizing factor” (EDHF) or “endothelium-derived relaxing factor” (EDRF), which has been a hot topic in vascular physiology and pathophysiology in the last two decades (Busse et al., 2002; Jiang et al., 2005). It is now known that the EDHF is often a variable combination of gap junction coupling, endothelial release of K+, nitric oxide (NO), epoxyeicosatrienoic acids (EETs) and prostanoid in various vascular beds (Griffith, 2004; Jiang et al., 2005). In guinea pig cochlear SMA, we demonstrated that ACh primarily stimulates the intermediate conductance KCa (IK) in endothelial cells (ECs) causing efflux of K+ ion and thus hyperpolarizing these cells (Guan et al., 2007; Jiang et al., 2007; Jiang et al., 2005). ACh-induced hyperpolarization in the VSMC is mainly (60%) an electrical spread of the EC-originated hyperpolarization via gap junctions; whereas, the K+ release from the EC activates an inward rectifier potassium channel (Kir) and the Na-K-ATP pump, causing the remaining ~40% hyperpolarization of the VSMC.
On the other hand, up until now, the cellular and sub-cellular mechanisms of ACh-induced depolarization and contraction in vascular cells have received few studies and remain poorly understood, although extensive studies of a similar muscarinic action have been conducted on gastrointestinal smooth muscle cells (Albert et al., 2006a; Zholos, 2006). Among the most relevant studies, (Beny et al., 1988) reported that ACh at low concentrations caused a persistent endothelium-dependent relaxation and hyperpolarization in rabbit aorta, whereas ACh depolarized and contracted the VSMCs after removal of the endothelial layer. Based on the effects of several agonists and antagonists for muscarinic receptor subtypes on pre-contracted rabbit aortic rings, (Jaiswal et al., 1991) concluded that the relaxation elicited by cholinergic stimuli in endothelium-intact preparations was mediated by activation of M3 receptors located on endothelial cells and the ensuing release of EDRF, while the contraction in endothelium-denuded aortic rings was mediated via stimulation of M2 receptors located on smooth muscle cells. Inoue et al., (2001) demonstrated that carbachol or ATP activated a cation current in HEK293 cells that expressed a transient receptor potential (TRP) channel TRPC6, which mimicked the cation current activated by α-adrenergic and muscarinic stimulation in dispersed rabbit portal vein smooth muscle cells. However, it remains unclear whether these observations can be extended to arterial cells in more physiological conditions.
In this study, using conventional intracellular recording and whole-cell tight seal recording techniques on in vitro guinea pig SMA preparations, we demonstrate that two ionic mechanisms, i.e., opening of a TRP-like cation channel and closure of a potassium channel, mediate the ACh-depolarization.
2. Materials and Methods
2.1. Animals and in vitro arterial preparation
The SMA segments were prepared as previously described (Jiang et al., 1999; Jiang et al., 2001). Briefly, guinea pigs (250 – 500 g) were anesthetized by intramuscular injection of an anesthetic mixture (1 ml/kg) of ketamine 500 mg, xylazine 20 mg and acepromazine 10 mg in 8.5 ml H2O and then killed by exsanguination. Both bullae were rapidly removed and transferred to a Petri dish filled with a physiological solution (Krebs) composed of (in mM): NaCl 125, KCl 5, CaCl2 1.6, MgCl2 1.2, NaH2PO4 1.2, NaHCO3 20, glucose 8.3, and saturated with 95% O2 and 5% CO2 at 35 °C (pH 7.4). The SMA was dissected out from the cochlea under a stereomicroscope. The vessels were incubated for 0.5 – 24 h in the Krebs solution and then transferred to a bath chamber for intracellular recording, or were enzyme-treated for whole-cell recording. The procedures were approved by the OHSU Animal Care and Use Committee.
2.2. Intracellular recording
A 2–5 mm long segment of the SMA, 40–80 μm outside diameter (OD), was pinned with minimum stretch to the silicon rubber layer (Sylgard 184, Dow Corning) in the bottom of the recording bath chamber (volume 0.5 ml) and continuously superfused with the Krebs solution at 35 °C. The outside connective tissue was further cleaned manually under a stereo-microscope (Nikon SMZ-2T). The glass microelectrode was filled with 2 M KCl and had a resistance of 60 – 150 MΩ. Intracellular penetration was obtained by advancing the electrode into the adventitial surface of the vessel with a micromanipulator (Narishige, MP-1, Japan). Transmembrane potential and injected current were simultaneously monitored with an NPI preamplifier (NPI, SEC10-LX, Germany). The electrical signals were recorded with a computer equipped with pClamp8 software (Axon Instruments, Inc.) using sampling intervals of 0.1, 0.5 or 10 ms. The resting potential was normally determined 5 min after the initial voltage jump at the penetration and checked by the voltage jump at the withdrawal of the electrode.
The input resistance was measured by applying 0.2–0.5 nA, 0.5–2 s current pulses via the recording electrode with the capacitance compensation and bridge-balance well-adjusted on the NPI preamplifier (Jiang et al., 2005). The adjustment was achieved by simultaneously using an additional data acquisition computer with a monitor displaying fast sweeps (0.5–2 s, Fig. 3B&D) of current and voltage signals at a 10 kHz sampling rate. In addition, 5 or 10 sweeps were averaged to minimize the baseline noise (Jiang et al., 2005).
Fig. 3. ACh-depolarization is associated with an increase or no change in input resistance in low Vm cells.

A&C: Chart recordings of membrane potentials of two different VSMCs. The input resistance (Rin) was monitored by electrotonic potentials (regular downward deflexions) induced by current pulses (I in B & D). B&D are high-speed recordings of the induced tonic potentials (V) simultaneously taken at times indicated at a–e in A and a–d in C, each being an average of five sweeps. Note that, in cell A, the high K+-induced depolarization was associated with a decrease in Rin (from 13.3 to 11.1 MΩ) whereas ACh-depolarization was associated with an increase in Rin (from 12.6 to 17.3 MΩ). In the presence of 18β-glycyrrhetinic acid (18βGA), the Rin increased to 34.5 MΩ during the ACh-depolarization from a pre-ACh control of 27.6 MΩ. In cell C, the Rin (18.5 MΩ at a) was decreased by high K+ (to 16.1 MΩ at b) and increased by 18βGA (to 28 MΩ at d) but was not significantly affected by ACh (18.5 MΩ, at c). All voltage scale bars indicate 10 mV.
Low K+ (nominal 0 mM) extracellular bathing solution was made by replacing KCl in the normal Krebs mentioned above with equimolar NaCl. High K+ (10 and 20 mM) extracellular solution was made by adding KCl and reducing NaCl accordingly. Low Na+ (20 mM) extracellular solution was made by replacing NaCl with equimolar NMDG (N-methyl-D-glucamine) from Krebs. When La3+ was administered, the normal Krebs was replaced by a HEPES buffer containing (in mM): NaCl 138, KCl 5, CaCl2 1.6, MgCl2 1.2, Na-HEPES 5, HEPES 6, glucose 8.3, and saturated with air at 35 °C (pH 7.4), to avoid precipitation.
2.3. Tight-seal whole-cell recording from VSMCs embedded in the SMA
The ACh-induced currents were also analyzed in VSMCs remaining embedded in SMA segments and briefly in a dissociated condition, as described in our previous study (Guan et al., 2007). A short segment (~0.3 mm long, ~35 μm OD) of the SMA was transferred to a 35-mm glass-bottomed Petri dish filled with aerated normal external solution composed of (in mM) NaCl 138, KCl 5, CaCl2 1.6, MgCl2 1.2, Na-HEPES 5, HEPES 6 and Glucose 7.5, with pH7.3 at room temperature. The preparation was secured at the bottom of the dish by the weight of a tiny platinum strip on each end and digested with collagenase A (0.75 mg/ml, Sigma) dissolved in the normal external solution at 36°C for 20–30 min. After a thorough rinse with the normal external solution, the vessel was further cleaned free of its adventitial tissue under a stereomicroscope. The SMA segment was then visualized under an inverted microscope (Zeiss, Axiovert 35) equipped with DIC function and continuously superfused with the normal external solution (0.2 ml/min) at room temperature (25°C). Glass recording pipettes had a tip about 1 μm OD and a resistance about 5 MΩ when filled with a normal internal solution containing (mM): K+-gluconate 130, NaCl 10, CaCl2 2, MgCl2 1.2, HEPES 10, ethylene glycol-bis [β-aminoethylether] N,N′,N′-tetraacetic acid (EGTA) 5 (118 nM free Ca2+) and glucose 7.5; adjusted to pH 7.2. By using a micromanipulator (Siskiyou Design Instruments, MX7600R & MX10L, USA), the glass micropipette tip was advanced and sealed onto the VSMC with a seal resistance between 1 and 20 GΩ. After a full compensation of the pipette capacitance, a strong negative pressure or a zap current was used to rupture the membrane, thus forming a whole-cell configuration. Membrane currents were low-pass filtered at 5 or 10 kHz (−3 dB). Data was recorded on a PC computer equipped with pClamp 9.2 software (Axon Instruments, Inc.) at a sampling interval of 10, 20 or 100 μs. A continuous chart recording was simultaneously conducted by a Minidigi digitizer (Axon Instruments, Inc. USA) at a sampling interval of 50 ms.
2.4. Drug application & statistics
Drugs of known concentrations were applied via the bathing solution. The solution that passed the recording chamber could be switched, without change in flow rate or temperature, to one that contained a drug(s) or one of different ionic composition. Drugs used in this study were: acetylcholine (ACh), 4-diphenylacetoxy-N-methylpiperidine methiodide (DAMP), 4-aminopyridine (4-AP), charybdotoxin (ChTX), nifedipine, nitrendipine (NTDP), diltiazem, linopirdine, glipizide, niflumic acid, indanyloxyacetic acid 94 (IAA-94) and 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS), flufenamic acid (FFA), amiloride (all from Sigma-Research Biochemicals Inc.) or 18β-glycyrrhetinic acid (18βGA, ICN, USA). The time of drug application was normally 100 s for ACh and >15 min for channel blockers except NTDP. The latter (1 μM) often took more than 30 min to exert a complete blockade of the ACh-hyperpolarization. Statistical values are expressed as means ± S.E.M.
3. RESULTS
3.1. General Observations
Conventional intracellular recordings and whole-cell recordings were made from more than 300 cells in 225 isolated SMA segments. The membrane properties of the cells obtained from intracellular recordings were similar to those reported previously and generally indistinguishable between the VSMC and EC (Jiang et al., 2001). Sampled cells normally showed an initial resting potential either near −40 or −75 mV, called high and low Vm, respectively. A cell may quickly shift its RP from a high Vm to a low Vm, or vice versa (Jiang et al., 2001). The sampling ratio of initial high to low Vm cells was 1.15 (n = 143/124) within 6 h after vessel isolation from the animal while more low Vm cells than high Vm cells were sampled after 24 h incubation (Jiang et al., 2001). When needed, the types of recorded cells (n = 30) were histologically and/or electrophysiologically identified as done in our previous studies (Jiang et al., 2005; Jiang et al., 2001). The sampling ratio of VSMCs to ECs in the present study was roughly 1:1 (Jiang et al., 2005; Jiang et al., 2001).
Whole-cell recordings were performed on more than 70 VSMCs embedded in SMA segments. The membrane properties were as described in our previous report (Guan et al., 2007). With basic internal solution and HEPES-buffered external solution, the input resistance of the embedded VSMCs at −40 mV was about 200 MΩ but increased to 2.2 GΩ by application of the gap junction blocker 30 μM 18βGA, resulting in a single exponential capacitive current relaxation and an input capacitance of ~7 pF, which indicates a complete electrical isolation of the recorded cell from its surrounding ones (Guan et al., 2007). The whole-cell current I/V curve often showed an obvious outward rectification upon depolarization beyond −30 mV and an inward rectification upon hyperpolarization beyond −60 mV (Fig. 5B, also see Guan et al., 2007), suggesting the expression of both delayed rectifier (KDR) and inward rectifier K+-channels (Kir) in the VSMCs.
Fig. 5. Whole-cell recording of ACh-induced current from VSMCs in SMA segments.
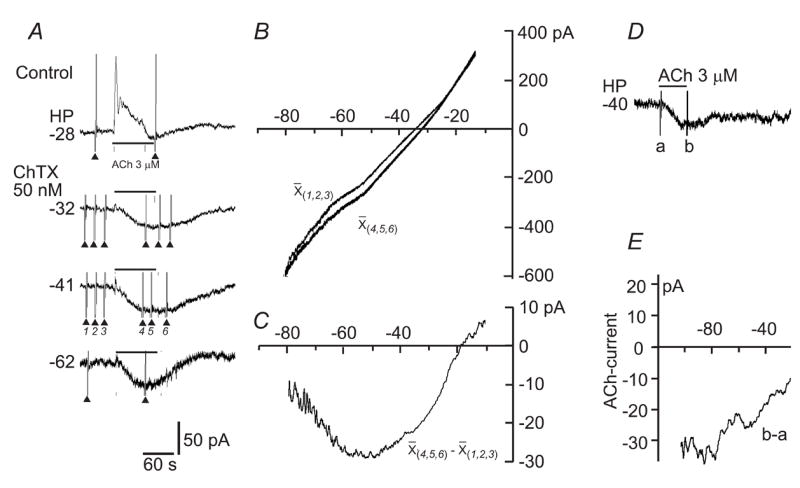
A: Chart traces of the whole-cell currents at different holding potentials (HP) showing that ChTX blocked ACh-induced outward current (Guan et al., 2007) and unmasked a larger inward current. The fast current deflexions (▴) were currents induced by ramp voltage commands (−120 to 0 mV). B: Ramp-command-constructed I/V curves before (X(1,2,3)) and during (X(4,5,6)) the ACh-current, each being an average of three repeats at the HP of −41 mV. The access resistance was compensated. C: The net ACh-current I/V curve, derived from curves in B, exhibited a U-shape I/V relation with two reversal potentials at −88 mV (with extrapolation) and −18 mV. D & E: AChinward current in another VSMC in the presence of 1 μM nitrendipine. Note that the I/V curve of the ACh-current showed a largely linear relation with a positive slope and a reversal potential of −0.4 mV. These U- and /-shaped I/V trajectory types were seen in 50% and 35% of total cells sampled, respectively. The remaining 15% cells showed a \shaped I/V curve (data not shown).
3.2. ACh causes dual membrane actions via muscarinic receptor(s) in the SMA
Bath application of ACh (3 –10 μM) caused a hyperpolarization, a depolarization, or a biphasic response, i.e., a hyperpolarization followed by a depolarization, depending on the pre-drug Vm level of the cell in nearly all SMA cells (Fig. 1A). ACh only induced a depolarization in cells that initially had a low Vm (~−75 mV) or had been shifted to that level, whereas it caused a robust hyperpolarization in cells that had a high Vm (~−40 mV) or had been depolarized to this level by Ba2+. ACh often induced a biphasic response, a hyperpolarization followed by a depolarization in intermediate Vm cells (~−60 mV, n =20, data not shown).
Fig. 1. ACh induces both depolarization and hyperpolarization in a cell of the spiral modiolar artery via M-receptor(s).
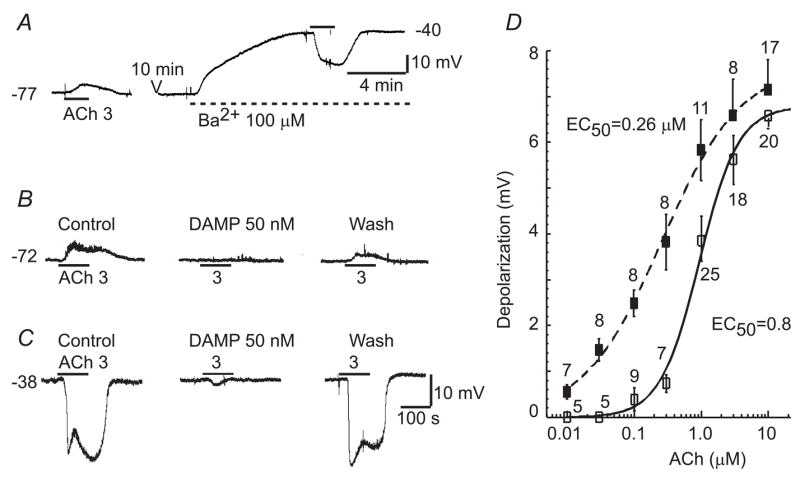
A: A sample chart record showing that 3 μM ACh depolarized the cell when it had an initial RP of −77 mV whereas ACh hyperpolarized the cell while it was depolarized to about −40 mV by the presence of Ba2+. B & C: Both ACh-depolarization and ACh-hyperpolarization were markedly blocked by a muscarine receptor (M3) antagonist 50 nM 4-DAMP, but they were sensitive to different channel blockers (see Fig. 3, (Jiang et al., 2005). The scale bars in C also apply to B. D: The ACh-depolarization and -hyperpolarization amplitudes were concentration-dependent. The data were all from low Vm (~−75 mV) cells. A Hill equation fit to the concentration-response data of ACh-depolarization (left curve) and - hyperpolarization (right curve) revealed an EC50 of 0.26 and 0.89 μM, respectively.
ChTX or NTDP, blockers for the intermediate conductance KCa, had no significant effect on the ACh-depolarization in the vast majority (91%) of low Vm cells tested (n ≥ 60, Fig. 2A&C), indicating that little ACh-hyperpolarization had contaminated the ACh-depolarization or had been masked by ACh-depolarization in these cases. In the remaining low Vm cells and nearly all intermediate Vm cells (n = 25), the KCa blockers significantly enhanced the ACh-depolarization, probably by two mechanisms, i.e., by removal of the hyperpolarization and, in some cases, by initiation of a regenerative Kir inactivation (Jiang et al., 2001). The KCa blockers, while suppressing the ACh-hyperpolarization, usually unmasked or enhanced the ACh-induced depolarization in cells that had a high Vm (Fig. 2B& D). The ACh-hyperpolarization is endothelium-dependent and will only be briefly mentioned for comparison since it has been thoroughly studied previously by us in the SMA (Guan et al., 2007; Jiang et al., 2007; Jiang et al., 2005) and by others in other vessel preparations (Griffith, 2004). Taking advantage of the fact that the ACh-hyperpolarization can be completely suppressed by 50 nM ChTX or 1 μM nitrendipine (NTDP) in the SMA (Figs. 2, 5, and Jiang et al., 2007), we were able to study the ACh-depolarization or the ACh-induced depolarizing current in isolation with the KCa-blocker present throughout the experiments. NTDP was more often used because it is also a known L-type Ca2+-channel (IL) blocker, which would prevent a secondary activation of the IL by the ACh-depolarization.
Fig. 2. Calcium-activated potassium channel blockers suppress ACh-hyperpolarization but not ACh-depolarization.

Both charybdotoxin (ChTX), a blocker selective for large conductance (BK) and intermediate conductance (IK) Ca2+-activated K+ channels (Van Renterghem et al., 1995), and nitrendipine, a blocker selective for IK, had no significant effects on ACh-induced depolarization in low Vm cells (A&C) while both compounds effectively suppressed the ACh-hyperpolarization in high Vm cells (B&D, also (Jiang et al., 2007). Note that, in B & D, after blockade of ACh-hyperpolarization by the KCa channel blockers, a small depolarization was unmasked. The voltage scale bar applies to all and the time scale label 100 s applies also to A–C.
Moreover, the ACh-depolarization was not significantly changed by the gap junction blocker 30 μM 18βGA in both the VSMC and the EC of the SMA (n = 6; Jiang et al., 2001), suggesting that it was generated in both the VSMC and the EC. We do not distinguish the response between these two cell types in this report unless noted otherwise.
Both ACh-depolarization and hyperpolarization were almost completely and reversibly blocked by 50 nM 4-DAMP, an antagonist relatively selective for M3 muscarinic receptor (n ≥ 10, not shown). The amplitudes of both the ACh-depolarization and hyperpolarization were concentration-dependent (Fig. 1B) with significantly different EC50s (0.26 ± 0.041 μM vs. 0.89 ± 0.17 μM, respectively, p < 0.05), suggesting that ACh-depolarization is a receptor mediated membrane action distinct from the ACh-hyperpolarization.
3.3. ACh-depolarization is associated with multiple conductance changes
The input resistance (Rinput) change accompanying the ACh-depolarization was monitored in current-clamp mode of the NPI preamplifier, as described previously (Jiang et al., 2005). In cells with high Vm (~−40 mV), ACh-depolarization was always associated with a decrease in input resistance (from 15.6 ± 2.4 MΩ to 12.3 ± 2.1 MΩ, n= 6, p < 0.05, paired t-test; not shown). However, in low Vm cells, two typical patterns of change were found in different cells. First, as shown in Figure 3A&B, ACh-depolarization was associated with an increase in Rinput in the majority of low Vm cells tested (from 8.7 ± 1.4 MΩ to 13.1 ± 1.7 MΩ, n = 8 of 12 cells tested, p < 0.01, paired t-test; RP = −76.3 ± 2.2 mV). Second, as shown in Figure 3C&D, ACh-depolarization was associated with no detectable Rinput change (from 14.1 ± 2.9 MΩ to 14.3 ± 2.9 MΩ, n = 4, p > 0.05, paired t-test, RP = −66.2 ± 3.2 mV). In comparison, in the same cell of either case, 10 mM K+ caused a comparable depolarization that was always associated with a decrease in Rinput (from 12.3 ± 3.6 MΩ to 10.1 ± 2.9 MΩ, n = 6, p<0.05). In the presence of the gap junction blocker 18βGA, the Rinput increase accompanying the ACh-depolarization remained (Fig. 3A, n = 3) or became undetectable (n = 2, not shown). The 25 μM 18βGA alone always caused a substantial increase in Rinput and a 5–10 mV depolarization, as described previously (Fig. 3, Jiang et al., 2005). A manual clamp of Vm back to the control level by applying a current via the recording electrode was not feasible because the resistance of the sharp electrode for the intracellular recording was too high (often ~100 MΩ or higher) to pass enough current (see Discussion).
The effect of background Vm on ACh-depolarization was analyzed in two ways: 1) by using incremental concentrations of Ba2+ to depolarize the cell to various stable levels (Fig. 4A&B), and 2) or by plotting the ACh-depolarization amplitudes against the native RP values of randomly sampled cells (Fig. 4C). The majority of cells tested (n = 5 of 8) showed a decrease in the amplitude of ACh-depolarization upon Ba2+-induced depolarization (Fig. 4A). The remaining three cells showed an increase in ACh-depolarization amplitude upon moderate (from ~−85 to ~−50 mV) depolarization and then a decrease upon strong (up to −20 mV) depolarization (Fig. 4B). The voltage dependence was probably not due to a direct blocking action of Ba2+ on the ACh-depolarization, because 1 mM Ba2+ caused no reduction or even a 1–3 mV enhancement of the ACh-depolarization in half the cases (n = 3 of 6, supplemental figure sFig. 1) along with a small (2–5 mV) background depolarization in the high Vm cells. In addition, Ba2+-induced moderate depolarization enhanced ACh-depolarization in some cells (Fig. 4B).
Fig. 4. Effects of membrane potential on ACh-depolarization.
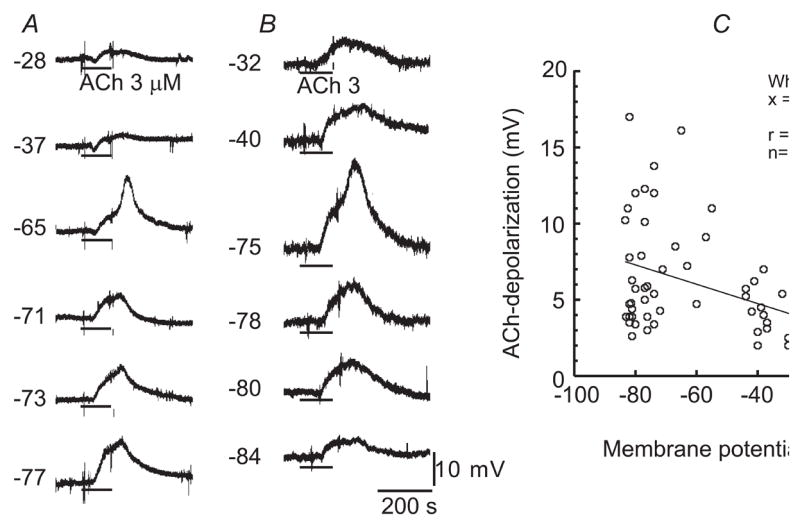
A&B: Membrane potential traces of two different cells, showing that the amplitude of ACh-induced depolarization was reduced in A, but was enhanced and then reduced in B upon membrane depolarization from an initial RP −77 and −84 mV, respectively. Note that, in the cases of Vm −65 mV in A and −75 mV in B, there was a spike-like depolarization riding on the smooth ACh-depolarization, the amplitude of which was disregarded. The background depolarization was induced by Ba2+ of incremental concentrations (0.1, 1, 10, 100 & 1000 μM). Nitrendipine (1 μM) was present throughout. C: Plots of ACh-depolarization amplitude against membrane potential of all the cells sampled in the presence of nitrendipine, but not Ba2+, showing an approximately negative slope linear relation.
The plots of ACh-depolarization amplitude against the native RP of 58 cells were quite scattered (Fig. 4C). A rough tendency was revealed by a least square fit to a linear function: A negative slope indicates that the ACh-depolarization became smaller in depolarized cells, despite the poor regression coefficient value of 0.38.
Taken together, the data obtained above suggested that a multiple, rather than a single ion conductance may be involved in the electrogenesis of ACh-depolarization.
3.4. I/V relation of ACh-induced inward current suggests activation of a cation current and inactivation of a potassium current
In the absence of a gap junction blocker, the capacitive relaxation of step command-induced currents in embedded VSMCs always followed a 3-term exponential function (Guan et al., 2007), indicating the charge/discharge process of multiple cells. Using the capacitive transient area (charge quantity) divided by step amplitude (mV), we estimated that the involved capacitance was 142 ± 27.9 pF (n = 19), which was about 23 times the value (6.1 pF) for the recorded cell chemically isolated by 30 μM 18βGA (Guan et al., 2007).
Figure 5A&B depicts measurements of a typical I/V relation of the ACh-induced current from a SMA muscle cell. ACh-induced current in embedded VSMCs appeared in three patterns: an outward current in 31 of 53 cells (58.5%, also Guan et al., 2007), an outward current followed by an inward current in 10 of 53 cells (18.9%, Fig. 5A), and an inward current only in 1 of 53 cells (1.9%). The remaining 11 of 53 cells showed no current response. When the ACh-induced outward current was completely suppressed by ChTX or 1 μM nitrendipine, an inward current was typically unmasked or enhanced (Fig. 5A).
By subtracting the control whole-cell I/V curve from the I/V curve during ACh-induced response, we constructed I/V curves of ACh-induced net inward current. The I/V curves showed three types of trajectories: back slash-shaped (\, data not shown), U-shaped (Fig. 5C) and forward slash-shaped (/, Fig. 5E) in 4, 13, and 9 cells, respectively, of the total 26 cells sampled. The U-shaped I/V curve had a negative slope segment from −120 to ~−60 mV and a positive slope segment from ~−50 to ~40 mV, with a reversal potential at −108 ± 2,8 mV and 1.5 ± 3.5 mV, respectively (curves being extrapolated in five cases). The flat segment between these two slope arms had a maximum amplitude of inward current (32.7 ± 5.4 pA), the voltage range of which varied from cell to cell, spanning a few mV to 40 mV between −70 to −30 mV. The \- and /-shaped I/V curves had a reversal potential of −85 ± 16.1 mV and −4.1 ± 3.5 mV, respectively.
We were able to construct the I/V curve of ACh-induced inward current only in two VSMCs in the SMA segment while the cell was completely isolated by the gap junction blocker 30 μM 18βGA (Guan et al., 2007). The ACh-induced current was small (−2.3 and −1.8 pA) at a holding potential of −40 mV, at which the baseline noise was normally minimized. One cell with a reasonable signal/noise ratio after averaging three repeats, showed an approximately linear I/V curve with a positive slope and a reversal potential of −13.6 mV (Fig. 6B). Similar experiments with dispersed VSMCs failed in a number of trials (n > 6). In all likelihood, this was probably due to the fact that the ACh-induced current was too small to be confidently resolved from the baseline noise (see Technical Issues in Discussion).
Fig. 6. I/V relation of ACh-induced inward current in an embedded VSMC isolated by 18βGA.

A: Gap-free trace showing ramp voltage command-induced currents before (a, 3 repeats) and during (b, 3 repeats) application of 3 μM ACh. Note that ACh caused a small inward shift of holding current (HC, −2.3 pA). B: I/V curve of the ACh-induced net current (b - a) was approximately linear. A least square fit to a linear function (dashed line) revealed a reversal potential of −13.6 mV. The curve was 3 Hz low-pass filtered.
3.5. Effects of extracellular Na+ and K+ concentration changes
Low Na+ (20 mM) caused a significant inhibition of ACh-depolarization in both low Vm and high Vm cells (Fig. 7). The inhibition amounted to 45.7 ± 6.2% (n = 4, Vm = −74 ± 2.0 mV) and 32.9 ± 4.2% (n = 6, Vm = −40 ± 1.1), respectively. Low Na+ alone generally induced a transient 3–5 mV depolarization followed by a 3–7 mV hyperpolarization upon wash-in and a reversed membrane response upon washout at both background Vm levels (Fig. 7A&B). The ACh tests were done at similar background Vm levels between the control and the low Na+ condition.
Fig. 7. ACh-depolarization is inhibited by alteration of extracellular Na+ and K+ concentrations.

A and B: Representative chart traces show that a low Na+ solution caused a ~46% depression of ACh-depolarization in low Vm cells (B) and a smaller inhibition (~33%) in high Vm cells (A). C: Low K+ (nominal 0 mM K+) and high K+ (20 mM) solutions both caused a ~45% reduction of ACh-depolarization in high Vm cells. NTDP (1 μM) was present in all cases, while 100 μM Ba2+ was present in all high Vm cells. Note that under these conditions, low Na+ alone induced a transient depolarization followed by a hyperpolarization in both the high & low Vm cells; zero K+ caused a few mV depolarizations in high Vm cells whereas 20 mM K+ produced a transient hyperpolarization followed by a recovery to a Vm near the control in all high Vm cells. Both the low and high K+ solution caused a large depolarization in low RP cells (not shown, see text). D: Column graph depicts the statistics of these effects shown in A–C. *,<0.05, **, p<0.01; paired t-test.
In high Vm cells, a nominal K+-free solution suppressed the ACh-depolarization by 44.7 ± 1.6%, and the high K+ (20 mM) solution reduced the ACh-depolarization amplitude by 44.5 ± 0.59% (Fig. 7). In cells of this Vm level with 100 μM Ba2+ present, the K+-free solution alone normally caused a 2–6 mV depolarization upon wash-in and a large (44–55 mV) transient hyperpolarization upon washout with normal Krebs, but the Vm always returned to the control level in 20–25 min (Fig. 7C). The high K+ solution itself normally produced a brief 10–20 mV hyperpolarization followed by a slow recovery to a level near the control Vm in a few min (Fig. 7C, also Jiang et al., 2001). High K+ washout also induced a transient hyperpolarization but the Vm quickly returned to the control level (Fig. 7C).
The nominal K+-free solution normally caused a 30 – 40 mV depolarization in low Vm cells, which was virtually a Vm shift from the low Vm (~−75 mV) to the high Vm (~−40 mV). This shift was probably due to an inhibition of Kir current since it is known that Kir conductance is extracellular K+-dependent and also that 100 μM Ba2+ mimics the action of zero K+ (Jiang et al., 2001; Quayle et al., 1996). The high K+ (20 mM) solution also caused a 20 – 30 mV persistent depolarization in such cells, owing to the change of EK value (Jiang et al., 2001). Therefore, it was not feasible to determine the effect of the K+ gradient change on ACh-depolarization at a similar low Vm level.
3.6. ACh-depolarization was distinctly inhibited by La3+, FFA and 4-AP but not by blockers for Ca2+-channels, Cl−-channels, KATP, IM and ENaC channels
Identifying blockers for the ACh-depolarization should offer other important evidence to clarify its ionic mechanism. Several blockers for non-selective cation channels, K+-channels and Cl−-channels were tested on the ACh-depolarization. We found that, in HEPES buffer, 1 mM La3+, the blocker for non-selective cation channels (NSCCs) including some TRP channels (Clapham, 2007; Manabe et al., 1995), effectively but not completely suppressed ACh-depolarization (Fig. 8) in both high Vm cells (by 42.4 ± 5.3%) and low Vm cells (by 48.3 ± 6.2%). The suppression was partially reversible after 30 min washout. La3+ alone often caused a small (2–3 mV) depolarization in high Vm cells and hyperpolarization in low Vm cells. HEPES buffer alone usually caused a 0.7 to 2 mV depolarization in high Vm cells and a 1.5 to 4 mV depolarization in low Vm cells (n ≥ 10), but caused no significant change in the ACh-induced depolarization (5.5 ± 0.48 from a control of 5.8 ± 0.84 mV in normal Krebs, n = 9 , p = 0.58, paired t-test). Of note, 1 mM La3+ also significantly suppressed ACh-hyperpolarization in cells of high Vm in the absence of IK blocker (by 80 ± 5.5%, from −15.5 ± 1.7 mV to −2.77 ± 0.44 mV, n = 8, p<0.05, not shown), which is consistent with a report that La3+ also inhibits store-depletion-triggered Ca2+ influx (Camello et al., 1999), and that the IK-mediated ACh-hyperpolarization apparently depends on this Ca2+ influx (Jiang et al., 2007). The weaker La3+ inhibition on ACh-depolarization than on ACh-hyperpolarization (p<0.05, Student’s t-test) suggests that the blocking mechanisms of La3+ may be different between these two ACh actions. The ACh-induced hyperpolarization was not significantly changed by application of HEPES alone for 30 min (from −11.2 ± 0.85 mV to −9.4 ± 1.1 mV, n = 6, p > 0.05).
Fig. 8. La3+, 4-AP and FFA distinctly suppress ACh-depolarization.

A: Representative chart recordings showing that ACh-depolarization in high Vm cells (a –c) was inhibited by La3+ (a) but not significantly by 4-AP (b) and FFA (c). In low Vm cells (d–f), La3+, 4-AP and FFA all significantly inhibited the ACh-depolarization. Ba2+ (100 μM) was present in a-c while nitrendipine (1 μM) in all cases. The resting potential is indicated on the left of each case. B: Column graph showing statistics of the collective data. Open columns are the controls, and filled columns are treatments. The number of cells tested and S.E.M. bars are at the top of the column pairs. * p<0.05, ** p<0.01 (paired t-test). Note that the combined application of 4-AP and FFA caused an inhibition stronger than that by either 4-AP or FFA alone (see text).
4-AP (1 mM) inhibited ACh-depolarization (Fig. 8A.e) in low Vm cells by 43.9 ± 6.1% (Fig. 8B, p = 0.0032), but only slightly in high Vm cells (by 16.9 ± 12%, p = 0.12, Fig. 8Ab). 4-AP alone produced a similar (p = 0.12) small depolarization in both low Vm cells (1.5 ± 0.17 mV, n = 15, RP = −78 ± 1.2 mV) and high Vm cells (2.1 ± 0.48 mV, n = 7, RP =−41 ± 3.4 mV). In addition, a combined application of La3+ and 4-AP caused a stronger inhibition of the ACh-depolarization (by 77 ± 3.5%, or from 6.5 ± 0.78 mV to 1.41 ± 0.22 mV, n = 9 including 4 high Vm and 5 low Vm cells) (sFig. 3). 4-AP had no significant effect on ACh-hyperpolarization (Jiang et al., 2005).
FFA, an inhibitor for some TRP channels (Clapham, 2007) and chloride channel (Goto et al., 2007), inhibited ACh-depolarization in low Vm cells by 24.8 ± 4.67% but had no significant effect in high Vm cells (1.33 ± 1.44%) (Fig. 8). FFA (10 μM) alone caused a small hyperpolarization (1.8 ± 0.31 mV, n = 10, sFig. 4) in high Vm cells while it produced no significant Vm alteration in low Vm cells (n = 6). A combination of FFA and 4-AP caused an enhanced inhibition on the ACh-depolarization (by 63 ± 18% in 16 low Vm cells and by 71 ± 22% in 5 high Vm cells). Interestingly, FFA (10 – 30 μM) significantly enhanced the ACh-hyperpolarization in the majority of high Vm cells sampled (from −11.5 to −15.6 mV, n = 9, p = 0.021, sFig. 4), maybe due to its specific inhibition on the ACh-depolarization.
On the other hand, the ACh-depolarization in both high and low Vm cells was not significantly affected by: 1) the blockers for ATP-sensitive K+-channel 3 μM glipizide, and the calcium-activated K+-channel 50 nM ChTX and 1 μM NTDP (Figs. 2, 5 & 8), 2) the chloride channel blockers 10 to 100 μM niflumic acid, IAA-94 and DIDS (sFig. 3), 3) the voltage-gated Ca2+-channel blockers 100 μM Cd2+, Ni2+, 10 μM nifedipine and 10 μM diltiazem, 4) the M-channel blocker 10 μM linopirdine (sFig. 3), (n ≥ 6 in each case, p > 0.05), and 5) the epithelial sodium channel (ENaC) blocker 10 μM amiloride in both high and low Vm cells (n ≥ 4, not shown).
4. Discussion
To the best of our knowledge, the present study is the first to analyze the ionic mechanisms underlying the ACh-induced depolarization in arteriolar cells using intracellular and whole-cell recording methods. Our main finding is that the ACh-induced depolarization via muscarinic receptors is generated by modulation of at least two distinct conductances, activation of a TRP-like non-selective cation conductance and inactivation of a potassium conductance, possibly of the Kir channel. Our study extended in several aspects the previous finding that a TRPC6-like NSCC is activated by muscarinic stimulation in dispersed portal vein smooth muscle cells (Inoue et al., 2001). First, that muscarinic stimulation activates a NSCC is also true in arterial cells under more physiological conditions, i.e., remaining in an acutely isolated vascular segment. Second, the pharmacological antagonism observed is not fully consistent with the feature of TRPC6, indicating that a different channel isoform(s) may be implicated in the molecular identity of the channel (see discussion below). Third, the ACh-depolarization was sensitive to 4-AP and the whole-cell current showed a reversal potential near EK, both suggesting that inactivation of a K+-conductance also contributes to the ACh-depolarization at least in a portion of the vascular cells (see below).
4.1. Technical issues
The paucity of reports in the literature on membrane mechanisms of vascular ACh-depolarization may be due to a number of causes. First, in most vascular tissues, a robust ACh-induced endothelium-dependent hyperpolarization often masks or seriously contaminates the ACh-depolarization, which would have complicated the study (Figs. 1 & 2). The pharmacological isolation of ACh-depolarization by a complete blockade of the KCa-mediated ACh-hyperpolarization has only recently become available in certain vessel preparations such as the in vitro SMA, where the cellular and ionic mechanisms of the ACh-hyperpolarization have become well understood (Jiang et al., 2005), and the affordable IK blocking agents have been identified (Jiang et al., 2007).
Second, the vascular ACh-depolarization itself is complicated regarding its cellular origin and its ionic mechanism. As we have shown above, the ACh-depolarization was generated in both VSMCs and ECs and it involved more than one ion conductance, at least in the guinea pig SMA. In the present study, we recorded single cells that typically remained gap junction-coupled. Therefore, it remains to be verified whether the mechanisms identified apply to either or both of the cell types. Figure 6 showed that ACh-current had a /-shaped I/V curve in the isolated VSMC, suggesting at least the cation conductance activation occurred in the VSMC.
Third, there is no known channel blocker that specifically blocks ACh-depolarization. Identification of a specific channel blocker(s) often greatly helps determine the ionic mechanism(s) of the membrane action of a neurotransmitter or a vasoactive agent (e.g., Si et al., 2002). The near complete blockade of ACh-depolarization by a combination of La3+ and 4-AP is consistent with involvement of two conductances. However, the channel nature of these conductances remains to be identified (see below).
Finally, it is indeed technically difficult to study the electrophysiological mechanism of the ACh-induced vascular depolarization. The sharp electrode suitable for conventional intracellular recording must be very fine in the tip, often with a resistance above 100 MΩ, which can pass a persistent current no larger than 0.5 nA and is unable to do significant Vm control (often < 5 mV) on the cell in the vessel segment (Hirst et al., 1989; Jiang et al., 1999). Thus, a manual clamp of the recorded cell to the desired Vm is not feasible, unlike the case of neurons (Jiang et al., 1981). When the Rinput measurements were done at two quite different Vm levels, the interpretation for the Rinput change could be compromised by the possible involvement of rectification properties of the cell. In our case (Fig. 3), we had to contrast the Rinput change of ACh-depolarization with that of a comparable depolarization by 10 mM K+. For the same reason, we had to use incremental elevations of Ba2+ concentration to depolarize the cells to analyze the effect of Vm on the ACh-depolarization (Fig. 4A&B). This method inevitably complicated the data interpretation so that more experiments (e.g., Fig. 4C and s Fig. 1) had to be performed to support the conclusion.
For whole-cell voltage-clamp experiments, now we know that the ACh-induced depolarizing current near the RP in a single VSMC is very small (often <2 pA) and difficult to be resolved from the baseline noise (Fig. 6), which is consistent with the report that the ACh-activated single channel has a very low open probability in physiological voltage range even with a very high agonist concentration (ACh 500 μM, Inoue et al., 1993). This is not surprising because the single VSMC has a high Rinput (3–10 GΩ in dispersed VSMCs, (Guan et al., 2007; Hirst et al., 1989; Wang et al., 1993)). According to Ohm’s law, a 2 pA current should roughly cause a 6–20 mV depolarization in cells of 3–10 GΩ, which is about the size of ACh-depolarization from intracellular recordings (Figs. 1 and 4C). In the present study, we circumambulated this problem by using voltage clamp of the VSMC remaining in a short arteriole segment in the absence of a gap junction blocker. The voltage step-elicited capacitive transient current exhibited a multiple term exponential relaxation and the input capacitance (Cinput) was estimated to be ~142 pF, all indicating that we were recording the whole-cell currents from multiple cells. Based on the fact that each embedded VSMC shows a Cinput of ~6.7 pF when it was isolated by 30 μM 18βGA (Guan et al., 2007), the 142 pF would be equivalent to approximately 20 cells that comprised the electrical circuit. Thus, the experimental approach could record ACh-induced current from multiple cells, which significantly enhanced the signal/noise ratio. In the mean time, we were aware that the space-clamping problem existed for the cells that surround the recorded one, so that the I/V relation curves obtained might be distorted more or less (Armstrong et al., 1992; Lindau et al., 1988). To minimize such distortion, we used the thinnest and shortest vessel segments possible for whole-cell recording. The reversal potentials for the K+-conductance inactivation and cation current activation by ACh were quite close to the equilibrium potentials for K+ (EK) and non-selective cation currents, suggesting that the space-clamping problem appeared tolerable in our cases.
4.2. The activation of a cation conductance
A line of evidence in this study supports that the ACh-depolarization implicates activation of a NSCC. This includes: 1) The ACh-depolarization was associated with a decrease in Rinput in all the cells with a high Vm. 2) In the majority of cells, the amplitude of ACh-depolarization was reduced upon membrane depolarization from low to high Vm. 3) The ACh-induced inward current showed an I/V curve that partially or entirely had a positive slope with a reversal potential near zero mV. 4) The ACh-depolarization was attenuated by a bathing solution with reduced Na+ or K+, which was expected to reduce the Na+ driving force or increase K+ driving force, respectively, thus attenuating the depolarization (Zholos, 2006). 5) ACh-depolarization was markedly suppressed by La3+, a known blocker for NSCC (Manabe et al., 1995). 6) ACh-depolarization was insensitive to voltage-gated Ca2+-channel blockers NTDP, Ni2+ and Cd2+, Cl−-channel blockers niflumic acid, IAA94 and DIDS, and ENaC blocker amiloride. The results excluded a possible role of a FFA-sensitive Ca2+-dependent Cl−-channel activation in the ACh-depolarization, as suggested in the mesenteric artery of spontaneously hypertensive rats (Goto et al., 2007).
It is not fully understood at the moment why the 20 mM K+ bathing solution also attenuated the ACh-depolarization. However, at least one mechanism would be a plausible reason. As high K+ induced an increase in Kir conductance (Jiang et al., 2001; Robertson et al., 1996), which was indicated by an initial transient large hyperpolarization followed by a small sustained hyperpolarization in high Vm cells (Fig. 7C, also (Jiang et al., 2001), the increased Kir conductance should exert a shunting effect on the ACh-induced current, causing a reduction of the depolarization.
The molecular identity of this ACh-activated NSCC would be important to clarify. With reference to literature information, our data of pharmacological antagonism and the feature of ACh-inward current are reminiscent of the transient potential receptor (TRP) channels (Clapham, 2007; Inoue et al., 2006; Zholos, 2006). Especifically, our data of ACh-depolarization or -inward current appears to share its features most with some TRPC channel sub-family members, particularly the TRPC3 and TRPC7, rather than TRPC6 as suggested for portal vein VSMCs (Inoue et al., 2001). It is known that more than 10 members in the TRP channel superfamily are expressed in the cardiovascular system and this number is growing (Inoue et al., 2006; Peppiatt-Wildman et al., 2007). Among the TRP members expressed in blood vessels, four of the seven TRPC subfamily members (TRPC1, 3, 6 and 7) form NSCC channels that are blocked by La3+ while TRPC4 and TRPC5 channels are activated by 100 μM La3+ (Albert et al., 2006b; Clapham, 2007). FFA blocks the cation currents mediated by TRPC3, 5, 7, TRPM4 and TRPP2 (Albert et al., 2006b; Clapham, 2007; Peppiatt-Wildman et al., 2007), but it potentiates the cation currents mediated by TRPC1 and TRPC6 (Hill et al., 2006; Inoue et al., 2001; Jung et al., 2002; Saleh et al., 2006). Since the ACh-depolarization presently studied was inhibited by both La3+ and FFA, involvement of homomeric channels of TRPC1, TRPC4, TRPC5 and TRPC6 seems unlikely. Furthermore, heteromeric channels within the TRPC subfamily, such as TRPC1/4/5 or TRPC3/6/7, have been observed in an expression system and in the native tissue, and their ion selectivity, pharmacology and gating property could be similar to or different from those of homomeric channels of its single isomers (Hofmann et al., 2002; Jung et al., 2002). It has been shown that the whole-cell current of TRPC6, not the TRPC3 or TRPC7, channels exhibited a U-shaped I/V relation, being similar to that of the ACh-current we observed (Fig. 5C, Albert et al., 2003; Albert et al., 2006b; Jung et al., 2002; Peppiatt-Wildman et al., 2007; Shi et al., 2004). In this regard, it is possible that TRPC6 may participate in the formation of a heteromeric NSCC channel that mediates the ACh-depolarization or inward current.
Taken together, although more pharmacological and molecular work are required to pinpoint the molecular identity of the NSCC responsible for the ACh-depolarization, our data suggest that the ACh-activated NSCC in this arteriole may involve a heteromeric TRPC channel(s) different from that in rabbit portal vein.
4.4. The inactivation of a potassium conductance
Evidence from this study suggests that inactivation of a potassium conductance contributes to the ACh-induced depolarization. First, in a portion of cells that had a low Vm near EK, ACh-depolarization was associated with an increase in Rinput (Fig. 3A) and a moderate depolarization from the highly negative Vm caused an increase in the amplitude of ACh-depolarization (Fig. 4B). Second, ACh-induced inward current had a U-shaped I/V curve in half the cells tested (Fig. 5A&B), and in many of these cases, the left arm of the curve reversed its polarity around EK. In contrast, the U-shaped whole-cell current I/V curves of TRPC6 or TRPC4 channels never showed a reversal potential at its left arm (Clapham, 2003; Jung et al., 2002). In a few cells, the I/V curve essentially had little or no right arm, suggesting that the inactivation of a K+-conductance played a dominant role in the electrogenesis in these cells. Third, the ACh-depolarization was inhibited by 4-AP, a known K+-channel blocker (Fig. 8), and 4-AP caused additional inhibition when added to a La3+-containing solution.
Several reports demonstrated that muscarinic stimulation inhibits inward rectifier K+-channels, e.g., in rat locus coeruleus neurons (Shen et al., 1992), in cardiac muscle cells (Dobrzynski et al., 2002) and in rat prefrontal cortex pyramidal neurons (Carr et al., 2007). Shen & North (Shen et al., 1992) presented a mathematical model of the two conductance alterations, activation of a cation conductance and inactivation of an inward rectifier K+-conductance, for the muscarine-induced depolarization in the neurons. The model reproduced a U-shaped I/V curve very similar to what we saw in Fig 5. Two other observations in this study do not appear supportive to a Kir inactivation. First, in cells with very negative Vm where Kir conductance was expected to be rather active, the presence of Ba2+ (0.1–1 μM) caused augmentation rather than inhibition of the ACh-depolarization in some cells (Fig. 4B). An argument could be made that such concentrations of Ba2+ blocked only a portion of Kir channels (indicated by a small depolarization, not a bimodal RP shift (Jiang et al., 2001), leaving enough Kir channels for ACh to act on. Second, 1 mM 4-AP is known as a selective blocker for a vascular delayed rectifier K+-channel (KDR or KV, Jackson, 2005; Nelson et al., 1995), not for Kir (Xu et al., 1999). We found that 4-AP markedly suppressed ACh-depolarization in the low Vm cells but only slightly in the high Vm cells, indicating that the 4-AP action was not likely to be related to its inhibition of KV, since KV is active only when Vm is more depolarized than −40 mV (Guan et al., 2007; Jackson, 2005). It is known that 1 mM 4-AP inhibits Kir by 10% in VSMCs (Nelson et al., 1995). It would be interesting to test whether 4-AP inhibits Kir in this particular preparation, or whether 4-AP can interfere with the interaction between ACh and the Kir channels.
In summary, using conventional intracellular and whole-cell recording techniques, we demonstrated that, when the EDHF mediated ACh-hyperpolarization was pharmacologically blocked, ACh induced a depolarization that was generated by activation of a TRP-like non-selective cation conductance and inactivation of a Kir-like potassium conductance in cochlear arteriolar cells. The results will help us to identify whether ACh participates in the neuromuscular transmission in the SMA and some other vascular beds. If a component of the evoked EJP shows the same ionic mechanism and mediated by the same receptor(s) like the ACh-depolarization, and if both the EJP and the ACh-depolarization can be enhanced by a cholinesterase inhibitor, the collective evidence will satisfy the criteria (Jiang et al., 1982; Snyder, 1980) for establishing a neurotransmitter role of ACh in the vascular regulation. It will also be interesting to determine whether ACh-mediated depolarization/vasoconstriction plays a role in generation of cerebral vasospasm, possibly with a companion cochlear vasospasm, in aneurysmal subarachnoid haemorrhage cases where the EDHF mechanism is compromised (Zhang et al., 1994), and to determine the effectiveness of cholinergic and ion channel interventions. .
Supplementary Material
Acknowledgments
The project described was supported by Grant Number R01 DC004716 (to ZGJ) from the National Institute On Deafness And Other Communication Disorders. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute On Deafness And Other Communication Disorders or the National Institutes of Health. This work was also supported by a grant from the Deafness Research Foundation (to ZGJ) and by the National Natural Science Foundation of China, No.30460043 (KTM). The authors are grateful to Lauren Grimm, Jill Lilly and Dr. Takatoshi Karasawa for reading the manuscript.
List of abbreviations
- ACh
acetylcholine
- 4-AP
4-aminopyridine
- Cinput
input capacitance
- ChTX
charybdotoxin
- DAMP
4-diphenylacetoxy-N-methylpiperidine methiodide
- DIDS
4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid
- EC
endothelial cell
- EDHF
endothelium-derived hyperpolarizing factor
- EGTA
ethylene glycol-bis [β-aminoethylether] N,N′,N′-tetraacetic acid
- EJP
excitatory junction potential
- EK
equilibrium potentials for K+
- FFA
flufenamic acid
- 18βGA
18β-glycyrrhetinic acid
- IAA94
indanyloxyacetic acid 94
- IK
intermediate conductance Ca2+-activated K+-channel
- IL
L-type Ca2+-channel or current
- KCa
calcium-activated potassium channel
- KDR or KV
delayed rectifier or voltage-gated K+-channel
- Kir
inward rectifier K+-channel
- NMDG
N-methyl-D-glucamine
- NSCC
non-selective cation channel
- NTDP
nitrendipine
- Rinput
input resistance
- SMA
spiral modiolar artery
- TRP
transient receptor potential channel
- Vm
membrane potential
- VSMC
vascular smooth muscle cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albert AP, Large WA. Signal transduction pathways and gating mechanisms of native TRP-like cation channels in vascular myocytes. J Physiol. 2006a;570:45–51. doi: 10.1113/jphysiol.2005.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Piper AS, Large WA. Properties of a constitutively active Ca2+-permeable non-selective cation channel in rabbit ear artery myocytes. J Physiol. 2003;549:143–56. doi: 10.1113/jphysiol.2002.038190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Pucovsky V, Prestwich SA, Large WA. TRPC3 properties of a native constitutively active Ca2+-permeable cation channel in rabbit ear artery myocytes. J Physiol. 2006b;571:361–9. doi: 10.1113/jphysiol.2005.102780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Gilly WF. Access resistance and space clamp problems associated with whole-cell patch clamping. Methods Enzymol. 1992;207:100–22. doi: 10.1016/0076-6879(92)07007-b. [DOI] [PubMed] [Google Scholar]
- Axelsson A. Comparative anatomy of cochlear blood vessels. Am J Otolaryngol. 1988;9:278–90. doi: 10.1016/s0196-0709(88)80036-x. [DOI] [PubMed] [Google Scholar]
- Baguley DM. Mechanisms of tinnitus. Br Med Bull. 2002;63:195–212. doi: 10.1093/bmb/63.1.195. [DOI] [PubMed] [Google Scholar]
- Beny JL, Brunet PC. Electrophysiological and mechanical effects of substance P and acetylcholine on rabbit aorta. J Physiol (Lond) 1988;398:277–89. doi: 10.1113/jphysiol.1988.sp017042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–80. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Camello C, Pariente JA, Salido GM, Camello PJ. Sequential activation of different Ca2+ entry pathways upon cholinergic stimulation in mouse pancreatic acinar cells. J Physiol. 1999;516(Pt 2):399–408. doi: 10.1111/j.1469-7793.1999.0399v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Surmeier DJ. M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J Neurophysiol. 2007;97:3432–8. doi: 10.1152/jn.00828.2006. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–24. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Clapham DE. SnapShot: mammalian TRP channels. Cell. 2007;129:220. doi: 10.1016/j.cell.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Dobrzynski H, Janvier NC, Leach R, Findlay JB, Boyett MR. Effects of ACh and adenosine mediated by Kir3.1 and Kir3.4 on ferret ventricular cells. Am J Physiol Heart Circ Physiol. 2002;283:H615–30. doi: 10.1152/ajpheart.00130.2002. [DOI] [PubMed] [Google Scholar]
- Goto K, Edwards FR, Hill CE. Depolarization evoked by acetylcholine in mesenteric arteries of hypertensive rats attenuates endothelium-dependent hyperpolarizing factor. J Hypertens. 2007;25:345–59. doi: 10.1097/HJH.0b013e328010d616. [DOI] [PubMed] [Google Scholar]
- Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan BC, Si JQ, Jiang ZG. Blockade of gap junction coupling by glycyrrhetinic acids in guinea pig cochlear artery: a whole-cell voltage- & current-clamp study. Br J Pharmacol. 2007;151:1049–1060. doi: 10.1038/sj.bjp.0707244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Hinton JM, Cheng H, Gao Z, Bates DO, Hancox JC, Langton PD, James AF. A TRPC-like non-selective cation current activated by alpha 1-adrenoceptors in rat mesenteric artery smooth muscle cells. Cell Calcium. 2006;40:29–40. doi: 10.1016/j.ceca.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Hirst GD, Edwards FR. Sympathetic neuroeffector transmission in arteries and arterioles. Physiol Rev. 1989;69:546–604. doi: 10.1152/physrev.1989.69.2.546. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–6. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultcrantz E. Clinical treatment of vascular inner ear diseases. Am J Otolaryngol. 1988;9:317–22. doi: 10.1016/s0196-0709(88)80039-5. [DOI] [PubMed] [Google Scholar]
- Inoue R, Kuriyama H. Dual regulation of cation-selective channels by muscarinic and alpha 1-adrenergic receptors in the rabbit portal vein. J Physiol. 1993;465:427–48. doi: 10.1113/jphysiol.1993.sp019685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res. 2006;99:119–31. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation channel. Circ Res. 2001;88:325–32. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Jackson W. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–27. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal N, Lambrecht G, Mutschler E, Tacke R, Malik KU. Pharmacological characterization of the vascular muscarinic receptors mediating relaxation and contraction in rabbit aorta. J Pharmacol Exp Ther. 1991;258:842–50. [PubMed] [Google Scholar]
- Jiang Z, Dun NJ, Karczmar AG. Substance P: a putative sensory transmitter in mammalian autonomic ganglia. Science. 1982;217:739–41. doi: 10.1126/science.6179162. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Dun NJ. Multiple conductance change associated with the slow excitatory potential in mammalian sympathetic neurons. Brain Res. 1981;229:203–8. doi: 10.1016/0006-8993(81)90758-7. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Shi X, Guan BC, Zhao H, Yang YQ. Dihydropyridines inhibit ACh-induced hyperpolarization in cochlear artery via blockade of intermediate conductance calcium activated potassium channels. J Pharmacol Exp Ther. 2007;320:544–551. doi: 10.1124/jpet.106.115212. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Nuttall AL, Zhao H, Dai CF, Guan BC, Si JQ, Yang YQ. Electrical coupling and release of K+ from endothelial cells co-mediate ACh-induced smooth muscle hyperpolarization in guinea-pig inner ear artery. J Physiol. 2005;564:475–87. doi: 10.1113/jphysiol.2004.080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZG, Qiu JH, Ren TY, Nuttall AL. Membrane properties and the excitatory junction potentials in smooth muscle cells of cochlea spiral modiolar artery in guinea pigs. Hear Res. 1999;138:171–180. doi: 10.1016/s0378-5955(99)00166-5. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Si JQ, Lasarev MR, Nuttall AL. Two resting potential levels regulated by inward rectifying potassium channels in guinea pig cochlea spiral modiolar artery. J Physiol (Lond) 2001;537:829–842. doi: 10.1111/j.1469-7793.2001.00829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Strotmann R, Schultz G, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–59. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 1988;411:137–46. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- Manabe K, Takano M, Noma A. Non-selective cation current of guinea-pig endocardial endothelial cells. J Physiol. 1995;487(Pt 2):407–19. doi: 10.1113/jphysiol.1995.sp020889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Nuttall AL. Sound-induced cochlear ischemia/hypoxia as a mechanism of hearing loss. Noise & Health. 1999;5:17–31. [PubMed] [Google Scholar]
- Peppiatt-Wildman CM, Albert AP, Saleh SN, Large WA. Endothelin-1 activates a Ca2+-permeable cation channel with TRPC3 and TRPC7 properties in rabbit coronary artery myocytes. J Physiol. 2007;580:755–64. doi: 10.1113/jphysiol.2006.126656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle JM, Dart C, Standen NB. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J Physiol (Lond) 1996;494:715–26. doi: 10.1113/jphysiol.1996.sp021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson BE, Bonev AD, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat coronary arteries: block by Mg2+, Ca2+, and Ba2+ Am J Physiol. 1996;271:H696–705. doi: 10.1152/ajpheart.1996.271.2.H696. [DOI] [PubMed] [Google Scholar]
- Saleh SN, Albert AP, Peppiatt CM, Large WA. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol. 2006;577:479–95. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF. Meniere's disease, pathogenesis and pathology. Am J Otolaryngol. 1982;3:349–52. doi: 10.1016/s0196-0709(82)80009-4. [DOI] [PubMed] [Google Scholar]
- Shen KZ, North RA. Muscarine increases cation conductance and decreases potassium conductance in rat locus coeruleus neurones. J Physiol. 1992;455:471–85. doi: 10.1113/jphysiol.1992.sp019312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–32. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si JQ, Zhao H, Yang Y, Jiang ZG, Nuttall AL. Nitric oxide induces hyperpolarization by opening ATP-sensitive K(+) channels in guinea pig spiral modiolar artery. Hear Res. 2002;171:167–176. doi: 10.1016/s0378-5955(02)00497-5. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Brain peptides as neurotransmitters. Science. 1980;209:976–83. doi: 10.1126/science.6157191. [DOI] [PubMed] [Google Scholar]
- Thalmann R, Miyoshi T, Thalmann I. The influence of ischemia upon the energy reserves of inner ear tissues. Laryngoscope. 1972;82:2249–72. doi: 10.1288/00005537-197212000-00013. [DOI] [PubMed] [Google Scholar]
- Van Renterghem C, Vigne P, Frelin C. A charybdotoxin-sensitive, Ca(2+)-activated K+ channel with inward rectifying properties in brain microvascular endothelial cells: properties and activation by endothelins. J Neurochem. 1995;65:1274–81. doi: 10.1046/j.1471-4159.1995.65031274.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mathers DA. Ca(2+)-dependent K+ channels of high conductance in smooth muscle cells isolated from rat cerebral arteries. J Physiol (Lond) 1993;462:529–45. doi: 10.1113/jphysiol.1993.sp019567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Rials SJ, Wu Y, Marinchak RA, Kowey PR. The properties of the inward rectifier potassium currents in rabbit coronary arterial smooth muscle cells. Pflugers Arch. 1999;438:187–94. doi: 10.1007/s004240050897. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cook D. Cerebral vascular smooth muscle potassium channels and their possible role in the management of vasospasm. Pharmacol Toxicol. 1994;75:327–36. doi: 10.1111/j.1600-0773.1994.tb00370.x. [DOI] [PubMed] [Google Scholar]
- Zholos AV. Regulation of TRP-like muscarinic cation current in gastrointestinal smooth muscle with special reference to PLC/InsP3/Ca2+ system. Acta Pharmacol Sin. 2006;27:833–42. doi: 10.1111/j.1745-7254.2006.00392.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.