

Abstract
Recent X-ray crystal structure determinations of Moloney murine leukemia virus reverse transcriptase (MoMLV RT) have allowed for more accurate structure/function comparisons to HIV-1 RT than were formerly possible. Previous biochemical studies of MoMLV RT in conjunction with knowledge of sequence homologies to HIV-1 RT and overall fold similarities to RTs in general, provided a foundation upon which to build. In addition, numerous crystal structures of the MoMLV RT fingers/palm subdomain had also shed light on one of the critical functions of the enzyme, specifically polymerization. Now in the advent of new structural information, more intricate examination of MoMLV RT in its entirety can be realized, and thus the comparisons with HIV-1 RT may be more critically elucidated. Here, we will review the similarities and differences between MoMLV RT and HIV-1 RT via structural analysis, and propose working models for the MoMLV RT based upon that information.
Keywords: reverse transcriptase, RNase H, Moloney Murine Leukemia Virus, Reverse transcription, retrovirus
1. Introduction
1.1. Reverse transcription
The reverse transcriptases (RTs) are a diverse group of enzymes, however they share two common functions: 1) a DNA polymerization function, which is capable of using either DNA or RNA as a template, and 2) an RNase H function, which serves to hydrolyze the RNA strand within an RNA/DNA hybrid. Both the polymerase and RNase H activities are essential for viral replication.
Reverse transcription is complex and results in the conversion of the viral RNA into dsDNA (for review, see (Abbink and Berkhout, 2008; Thomas and Gorelick, 2008)). Briefly, reverse transcription initiates from the 3’OH of a cellular tRNA, which is hybridized to the primer binding site (PBS) within the viral RNA genome. The specific tRNA primer varies between viruses, with MoMLV and HIV utilizing tRNAPro and tRNALys,3, respectively. RNA-primed-RNA-dependent DNA polymerization elongates the tRNA primer to the terminal 5’ repeat sequence (R) of the RNA, creating the first intermediate, the minus-strand strong-stop DNA ((−)ssDNA). Next, the RNase H must digest the RNA template to allow for the hybridization of the R regions duplicated at the 3’end of the (−)ssDNA and the 3’ end of the genomic RNA, resulting in the first strand transfer. Here the nascent DNA is then elongated via DNA-primed-RNA-dependent polymerization. Again the RNase H is recruited to hydrolyze the RNA template back to the hydrolysis-resistant polypurine tract (PPT), which subsequently serves as the new primer for second-strand DNA synthesis, using RNA-primed-DNA-dependent polymerization to elongate the PPT primer. RNase H is required to remove the PPT and tRNA primers. Removal of the tRNA primer results in the second-strand transfer via the interaction of the complementary PBS sequences. Finally, DNA-primed-DNA-dependent DNA polymerase completes the synthesis of the dsDNA, which retains the LTRs (U3-R-U5) at both of its ends, ready to be processed by the downstream integrase (IN).
1.2. Polymerase subdomains and RNase H
The structure of the E. coli Pol I Klenow fragment was found to fundamentally resemble the fingers, palm, and thumb of a right hand (Ollis et al., 1985), and this resemblance was extended to the description of the structural subdomains of the RTs. Thus the given subdomain designations in RT are (from N- to C-terminus): fingers (F), palm (P), thumb (T), connection (C); and the RNase H (R) domain confers multifunctionality to the RT. This anthropomorphic categorizing has turned out to be convenient in making reference to many of the structure/function activities of the RTs.
1.3. RT function
The multifunctional RTs possess two distinct active sites for the complex series of steps needed for proper reverse transcription. First, the polymerase active site, where all DNA elongation steps occur, is comprised of three conserved aspartate residues, and is located on a structurally conserved epitope in the palm subdomain. In addition to the catalytic aspartates, a conserved loop motif, Leu-Pro-Gln-Gly (residues 188–191 in MoMLV RT; residues 149–152 in HIV-1 RT; see Fig. 1), is thought to contribute to the proper positioning of the incoming dNTP with the substrate (Georgiadis et al., 1995). In the HIV-1 RT crystal structure of a ternary complex of RT:template/primer:dNTP, residues from the fingers subdomain, Lys65 and Arg72, are shown to coordinate the triphosphate moiety of the dNTP (Huang et al., 1998). The corresponding residues in MoMLV RT are Lys103 and Arg110, respectively. Second, the RNase H domain, critical for the hydrolysis of the RNA in general as well as for specific cleavages, is located C-terminally and has its active site composed of a conserved Asp-Glu-Asp motif. An additional aspartate residue, which is also conserved among RTs, is located extremely close to the C-terminus of the enzyme, and sometimes is included as part of an essential Asp-Glu-Asp-Asp quartet. RNase H activity has been defined as polymerase-dependent or independent (Furfine and Reardon, 1991), where in the polymerase-dependent mode, the 3’ terminus of the nascent DNA positions the RNase H for cleavage of the RNA template roughly 17–19 nucleotides downstream. Both the polymerase and RNase H active sites require specific divalent cations (Mg2+/ Mn2+) for optimal function. These two critical regions in HIV-1 RT and MoMLV RT have distinct similarities as well as characteristic differences.
Figure 1.
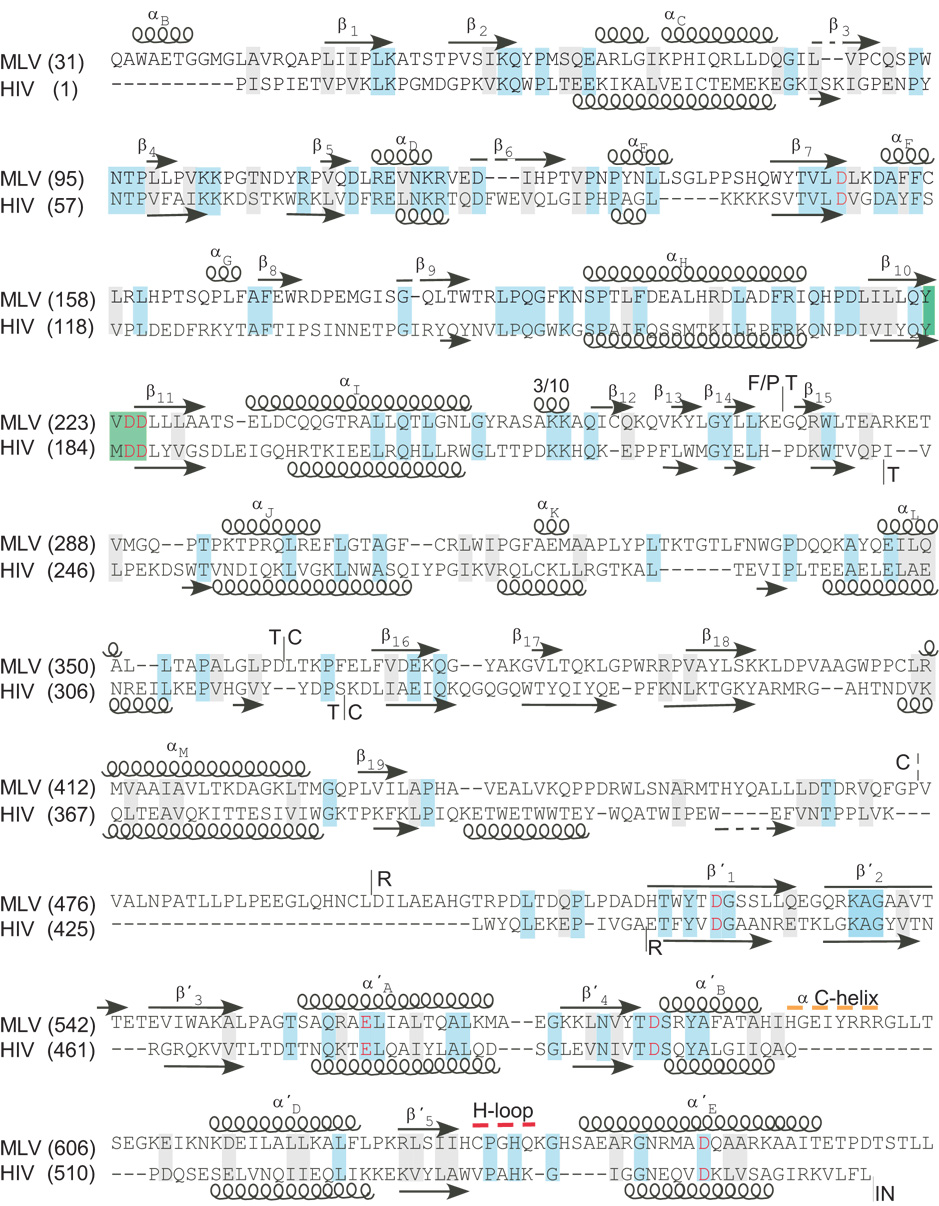
Sequence alignment and structural motifs of the MoMLV and HIV-1 RTs. Amino acid sequence of MoMLV from position 31–671 and HIV-1 RT from 1–562 are aligned based on sequence and structural motifs using the programs STRAP2 (Gille and Frömmel, 2001) and CE (Shindyalov and Bourne, 1998). Virus and amino acid position are indicated on the left. Positions of the α-helixes and β-sheets (arrows) are indicated above and below the sequences. Structural motifs of the polymerase domain are as previously defined (1RW3; (Das and Georgiadis, 2004)). Structural motifs of the RNase H domains are marked by a prime and are as previously defined (Lim et al., 2006). Sequence identities are highlighted in blue, conservative changes are highlighted in grey, the YXDD box is highlighted in green. Catalytic acidic amino acids are indicated in red. The putative α-C helix is overscored in a dotted orange line. The His-loop is overscored with a red dotted line. The boundaries of the F/P, T. C, and RNase H are shown with a vertical line. The broken vertical line on the MoMLV sequence indicates the position terminus of connection subdomain in the X-ray structure (1RW3).
1.4. HIV-1 RT structure
HIV-1 RT is one of the most extensively studied enzymes in the research community. In addition to the wealth of clinical, biochemical, and biophysical data amassed since the early 1980s, there also have been numerous published X-ray crystal structures of HIV-1 RT. The mature functional form of HIV-1 RT is a heterodimer, comprised of a 66-kDa, 560 amino acid subunit (p66), and a 51-kDa, 440 amino acid subunit (p51). The p66 subunit contains the fingers, palm, thumb, connection, and RNase H. The p51 subunit has the identical sequence of p66, however lacks the RNase H domain. The first structure of HIV-1 RT was in complex with the non-nucleoside reverse transcriptase inhibitor (NNRTI) Nevirapine, which in addition to confirming the functional heterodimer, imparted great insight into the flexibility of the subdomains of HIV-1 RT and their functions (Kohlstaedt et al., 1992).
The ensuing multitude of published crystal structures of HIV-1 RT have provided us with probative snapshots of the enzyme as it is working. Several representative structures are those of the unliganded apoenzyme (Esnouf et al., 1995; Hsiou et al., 1996; Rodgers et al., 1995), in complex with various NRTIs (Sarafianos et al., 2002; Tuske et al., 2004), in complex with various NNRTIs (Ding et al., 1995a; Ding et al., 1995b), in complex with dsDNA (Ding et al., 1998), in complex with an RNA/DNA hybrid (Sarafianos et al., 2001), and in a ternary complex with a dsDNA template/primer and incoming dNTP (Huang et al., 1998).
1.5 MoMLV RT structure
Prior to 2004 the only known crystal structures of MoMLV RT were those of the N-terminal fragment, the fingers/palm subdomain. In 1995 the first crystal structure of MoMLV RT fingers/palm was published, making it possible to begin structural comparisons with HIV-1 RT, principally in the polymerase active site region (Georgiadis et al., 1995). Several structures of the MoMLV RT fingers/palm domain in complex with various dsDNAs then followed, imparting greater insight into the structural invariance of the region, and detailing a unique (and conserved) binding site for blunt-ended DNA (Coté and Georgiadis, 2001; Coté et al., 2000; Najmudin et al., 2000). Biochemical studies had demonstrated that MoMLV RT was a functional monomer of 671 amino acids, whose subdomains consisted of fingers, palm, thumb, connection; along with the RNase H domain (Roth et al., 1985; Tanese and Goff, 1988). Absolute structural confirmation of MoMLV RT was achieved with the publication of the crystal structure of the full-length enzyme in complex with duplex DNA (PDB code 1RW3; (Das and Georgiadis, 2004)). In addition, the crystal structure of the isolated MoMLV RT RNase H domain was recently published (PDB code 2HB5; (Lim et al., 2006)). Although there are far fewer crystallographic snapshots of MoMLV RT, reasonable postulates as to its structure/function activity may be presented by comparing them with HIV-1 RT.
2. Sequence Comparisons of MoMLV RT and HIV-1 RT
It is well known that among the vast array of RTs in the scientific literature the overall sequence identities can be as meager as 10%, however the gross structural folds exhibited by these proteins are quite closely related (for a comprehensive review, see (Coffin et al., 1997)). In some respects it is a difficult task to compare the functional monomeric MoMLV RT with the functional heterodimeric HIV-1 RT, however with the knowledge of the full-length structure of MoMLV RT, the comparisons now can be further elucidated. Using a structure-based sequence alignment (see Fig. 1) in comparing the full length MoMLV RT with HIV-1 RT p66, it readily can be seen that the greatest sequence identity between the two enzymes resides in the fingers/palm subdomain, approaching 25% for the common 243 residues (see Fig. 1). If one allows for very conservative comparisons (e.g., Ala vs. Val, Leu vs. Ile, etc.), then the sequence identity rises to about 37%. The thumb subdomain, although retaining a similar overall fold (vide infra), has only a 12% sequence identity (79 residues). The connection subdomain, the region with the least sequence homology, has roughly 6% identically matched residues between the two enzymes. It is possible that this is due in part to the comparison being limited to that between MoMLV RT and HIV-1 RT p66 only. For example, in the crystal structure of HIV-1 RT in complex with an RNA/DNA hybrid (Sarafianos et al., 2001), it was shown that 3 residues from p51 comprise part of the RNase H primer grip or make contacts with the template, and thus present a “structural insertion” that cannot be described by a linear sequence comparison to p66 alone (see Table 1). In the RNase H domains, the exact sequence identity between MoMLV RT and HIV-1 RT increases back to about 20%, with conservative matches resulting in an agreement of approximately 30%. It should be kept in mind that directly comparing the monomeric MoMLV RT and the heterodimeric HIV-1 RT can present certain challenges; however, the publication of the two aforementioned MoMLV RT crystal structures allows greater confidence to do so.
Table 1.
Comparison of Selected Structural Motifs of MoMLV RT and HIV-1 RT
| Domain/Subdomain Motife | MoMLV RT residues | HIV-1 RT residuesa | |
|---|---|---|---|
| Fingers | 41–124; 160–192 | 1–84; 120–150 | |
| Palm | 1–40; 125–159; 193–275 | 85–119; 151–243 | |
| Fingers/Palm Thumb | 1–275 276–361 | 1–243 244–322 | |
| Connection | 362–496 | 323–437 | |
| RNase H | 497–671 | 438–560 | |
| Polymerase active site | Asp150, Asp224, Asp225 | Asp110, Asp185, Asp186 | |
| RNase H active site | Asp524, Glu562, Asp583, Asp653 | Asp443, Glu478, Asp498, Asp549 | |
| YXDD | Tyr222, Val223, Asp224, Asp225 | Tyr183, Met184, Asp185, Asp186 | |
| Polymerase Primer Grip | 267–274 | 228–235 | |
| MGBT | 295–318 | 255–268 | |
| RNase H Primer Grip | Val402b, Gly403b, Trp404b, Ser557, Ala558, Gln559, Arg560, Tyr586, Thr590 | p66:Gly359, Ala360, His361, Thr473, Asn474, Gln475, Lys476, Tyr501, Ile505 | |
| N/A c | p51:Lys395, Glu396 | ||
| RNase H Template Contactsd | Leu529, Ala558, Gln559, Arg585, His638 | p66:Arg448, Asn474, Gln475, Gln500, His539 | |
| N/Ac | p51:Lys390 | ||
| dNTP BP | Asp153, Phe155, Phe156, Gln190, Val223 | Asp113, Tyr115, Phe116, Gln151, Met184 | |
| NNRTI BP | N/A | 100 –110; 180 –190; 220 –240 | |
Domain/subdomain divisions taken from (Rodgers et al., 1995)
Not previously characterized, since the MoMLV connection subdomain structure was unknown
No p51 analog is known in this context
Some residues are in common with those of the primer grip
Abbreviations: MGBT = Minor groove Binding Tract;
dNTP BP = dNTP Binding Pocket;
NNRTI BP = NNRTI Binding Pocket;
Since MoMLV RT contains 671 amino acid residues, whereas HIV-1 RT p66 contains 560, it is obvious that there must be regions where there is no structural correspondence between the two enzymes. Referring to the structure-based sequence alignment between MoMLV RT and HIV-1 RT p66, three distinct regions account for most of the excess 111 residues present in MoMLV RT. Figure 1 shows where these three areas occur. First, and most distinctly, the immediate N-terminus of MoMLV RT contains 40 additional residues that are not present in HIV-1 RT. This renders the fingers/palm region of MoMLV RT substantially larger, however a superpositioning of the common structural motifs at the N-termini shows that the first 40 residues of MoMLV RT reside quite distant from the polymerase active site and the bulk of the fingers/palm in general (vide infra). Interestingly, the construct used for the full length MoMLV RT structure excludes the first 23 amino acids of the enzyme (Das and Georgiadis, 2004), which several studies had shown to be dispensable for its proper function (Das and Georgiadis, 2004; Das and Georgiadis, 2001; Gu et al., 2001). Second, in the region between the connection and RNase H, there are 32 additional amino acids in MoMLV RT. This may be a highly flexible region in MoMLV RT, and possibly could be formally unstructured (i.e., neither a β-strand nor an α-helix), since no electron density was observed for these residues in the crystal structure of the full-length enzyme (Das and Georgiadis, 2004). It must be noted here that in a linker-scanning analysis of this region (residues 475–502) in MoMLV RT, neither of the two isolates having an insertion in this region was viable in vivo, demonstrating that the region, whatever its secondary structure, is absolutely essential for viral function (Puglia et al., 2006). Also, linker insertions within this region were temperature-sensitive for RT activity in vitro (Tanese N, 1991). It is also quite possible that this region, seemingly quite solvent exposed in any of the proposed models, may be structurally critical for key movements of the RNase H domain over a wide range of distances. In addition, in the RNase H domain itself, there is a putative C-helix and loop region consisting of 11 residues not present in HIV-1 RT (see Fig. 1). It is noteworthy that this C-helix region was excluded in the crystal structure of the isolated RNase H domain of MoMLV RT (MoMLV RT RNase HΔC), since this motif is not seen in HIV-1 RT (see discussion below) (Lim et al., 2006). Thus, three specific regions of MoMLV RT account for roughly 76% of the excess residues found in MoMLV RT versus HIV-1 RT p66.
3. The Structural Subdomains of HIV-1 RT and MoMLV RT
HIV-1 RT is a functional heterodimer consisting of the p66 and p51 subunits whose sequences are identical except that p66 contains RNase H. The role of p51 has been shown to be largely structural, and the spatial orientations of its subdomains are quite distinct from those of p66, e.g., the polymerase active site of p51 is buried in the heterodimer (Kohlstaedt et al., 1992; Mulky et al., 2004; Rodgers et al., 1995). MoMLV is a functional monomer, with the four subdomains (F, P, T, and C) and the RNase H domain (R) comprising its structure. The N-terminal fingers/palm subdomains of MoMLV RT and HIV-1 RT are not sequentially distinct. In MoMLV RT there are three observed crossings of the polypeptide chain from the fingers to the palm. The N-terminal residues 24–41 reside in the palm subdomain, and the initial trajectory suggests that the preceding residues may also reside there. After residue 41 of MoMLV RT, the structural similarities with HIV-1 RT fingers/palm are quite evident, with two crossings of the polypeptide chain from the fingers to the palm (see Table 1). Oftentimes for ease of reference, the fingers and palm subdomains are collectively referred to as a single unit, the fingers/palm subdomain (i.e., F/P).
The polymerase active site is located within the palm subdomain, and contains three conserved aspartate residues residing on a β-sheet consisting of three antiparallel β-strands: β7, β10, and β11 in MoMLV RT and β6, β9, and β10 in HIV-1 RT (see Fig. 1). The three catalytic aspartates of the polymerase active site are Asp150, Asp224, and Asp225 in MoMLV RT, and Asp110, Asp185, and Asp186 in HIV-1 RT. Two of the aspartate residues in the polymerase active site are part of the conserved YXDD motif, which in MoMLV RT is YVDD, and in HIV-1 RT is YMDD. This structural motif, seen in all RTs, is unusual in that it involves a type II’ β hairpin, which generally favors glycine as the second residue of the turn, and neither MoMLV nor HIV-1 RT has a glycine at this position. Interestingly, one of the mutants in HIV-1 RT most resistant to clinical treatment for HIV-1 infection is Met184Val, the sequence analog seen in MoMLV RT (Boyer et al., 2007).
The thumb subdomain, the smallest of the five, contains two principal helices in both MoMLV RT and HIV-1 RT, and contains the minor groove binding tract (MGBT) (Bebenek et al., 1995) located on the α-helix facing the fingers subdomain. The connection subdomain, despite having the least formal sequence identity, has the same overall fold in the two enzymes, with similar residues serving as part of the RNase H primer grip: Val402, Gly405, Trp406 in MoMLV RT and Gly359, Ala360 and His361 in HIV-1 RT (see Fig. 1).
Despite its having slightly less sequence identity than that of the F/P subdomain, the RNase H domain is the most structurally similar region between the two proteins, and also exhibits remarkable overall fold similarity to other RNases H (Davies et al., 1991; Ishikawa et al., 1993; Katayanagi et al., 1992; Nowotny et al., 2005; Tsunaka et al., 2005). The RNase H domain contains the strictly conserved D-E-D motif whose residues are Asp524, Glu562, and Asp583 in MoMLV RT, and Asp443, Glu478, and Glu498 in HIV-1 RT. A fourth conserved aspartate, Asp653 in MoMLV RT and Asp549 in HIV-1 RT, is also important in RNase H activity. Table 1 presents a structural comparison of selected sequence motifs shared by MoMLV RT and HIV-1.
4. The full length MoMLV RT structure
The full-length MoMLV RT (PDB code 1RW3) was crystallized in a complex with DNA oligonucleotides having the sequence 5’-d(ATTGATATATTAAATT)-3’ and 5’-d(TAAATTTAATATATCA)-3’ resulting in a 14-mer duplex containing AT overhangs at each end (Das and Georgiadis, 2004). Unfortunately, the electron density was too ill defined for meaningful characterization of the nucleic acid; however, in the vicinity of the polymerase active site (residues Asp150, Asp224, and Asp225) electron density representing two base pairs from the dsDNA was observed. In addition, absolute characterization of the RNase H domain could not be achieved, due to insufficiently defined electron density. However, a general positioning of the RNase H domain was done manually, resulting in improved electron density upon further refinement, indicating that the overall location is correct (Das and Georgiadis, 2004). The exit of the connection subdomain in combination with the placement of the RNase H domain indicated that this structure of MoMLV RT was quite different from those observed in HIV-1 RT. It is precisely these caveats of 1RW3 - especially the missing residues 475–502 between the connection and RNase H - that prompted the creation and examination of additional models of MoMLV RT and how they may assist in profiling the function of the monomeric enzyme. In the ensuing discussions, the experimentally determined structure of the full length MoMLV RT will be referred to as Model 1. The generation of two additional models of the full length MoMLV RT is also discussed in detail below.
5. MoMLV RT RNase H
The high-resolution (1.6 Å) crystal structure of the isolated MoMLV RT RNase H domain was recently published (PDB code 2HB5, (Lim et al., 2006)). The construct crystallized lacks an 11 amino acid stretch, and is referred to as MoMLV RT RNase HΔC (RHΔC). Previous sequence alignments and modeling studies indicated that MoMLV RT RNase H and E. coli RNase H likely shared a common structural motif that is not seen in HIV-1, Bacillus halodurans (Bh), or avian retroviral RNases H (Davies et al., 1991; Leis et al., 1993; Nowotny et al., 2005). This motif observed in E. coli RNase H is an α-helix in the center of its structure, known as the C-helix, whose amino acid sequence is WIHNWKKR (residues 81–88 in PDB file 2RN2 (Katayanagi et al., 1990)). In a structure-based sequence alignment, the corresponding residues in MoMLV RT RNase H are HGEIYRRR (residues 594–601 in the full-length MoMLV RT; see Fig. 1). Because attempts to crystallize the wild type MoMLV RT RNase H domain did not yield crystals, the crystallization of the RHΔC mutant was attempted. Even though RHΔC retains little in vitro RNase H activity (Lim et al., 2002) and is incapable of in vivo replication (Boyer et al., 2001; Telesnitsky et al., 1992), it was pursued as a candidate for crystallization given that HIV-1 RT RNase H does not possess this structural motif (see Fig. 1).
In addition to the deletion of the putative C-helix from the construct, the RHΔC construct omitted the flanking residues Ile593, Gly602, and Leu603, such that in the PDB file (renumbered to reflect the full-length MoMLV RT) His592 is immediately followed by Leu604 (see Fig. 1). The high resolution of the data allowed for the fitting of several amino acids into two separate conformations, each at half-occupancy (see below).
Despite the high resolution of the data in 2HB5, there were three small regions, representing 15 amino acids, which had little or no electron density, suggesting a high degree of disorder. Two regions at the termini could not be characterized: six residues at the extreme N-terminus and four at the C-terminus. The third missing region, however, is a highly flexible solvent-exposed loop region, which contains His638 (see Fig. 1). This loop region was shown to contribute to proper substrate binding and positioning for RNase H activity in E. coli (Kanaya et al., 1991b), and was referred to as the “His-containing loop” within RT because the conserved histidine was necessary for viral reproduction (Tisdale et al., 1991). In the crystal structure of the isolated HIV-1 RT RNase H, this “His loop” with its conserved His539, was shown to be disordered (Davies et al., 1991), as it was in the solution NMR structures (Pari et al., 2003; Powers et al., 1991), and two of the mutant Bacillus halodurans crystal structures (Nowotny and Yang, 2006). Interestingly, in the 1HYS structure of HIV-1 RT in complex with the RNA/DNA hybrid, the His-loop and His539 are well characterized (see below).
6. Superpositionings of MoMLV RT and HIV-1 RT
6.1. HIV-1 has a mobile thumb subdomain
The numerous crystal structures of HIV-1 RT in a variety of experimental conditions have allowed for the observation of virtual freeze-frames of the enzyme at work. These snapshots illustrate the complexity of the hinging/swiveling motion of HIV-1 RT, which also has been discussed in many articles (Jager et al., 1994; Madrid et al., 2001; Sluis-Cremer et al., 2004; Temiz and Bahar, 2002), profiling the correlated/anticorrelated “ratcheting” action of a reverse transcriptase. For structural comparisons between MoMLV RT and HIV-1 RT, we considered three general categories into which we could place the HIV-1 RT structures. The categorization relies heavily upon the depiction of how the thumb subdomain is situated: “closed”, “open”, and “splayed” (Fig. 2).
Figure 2.
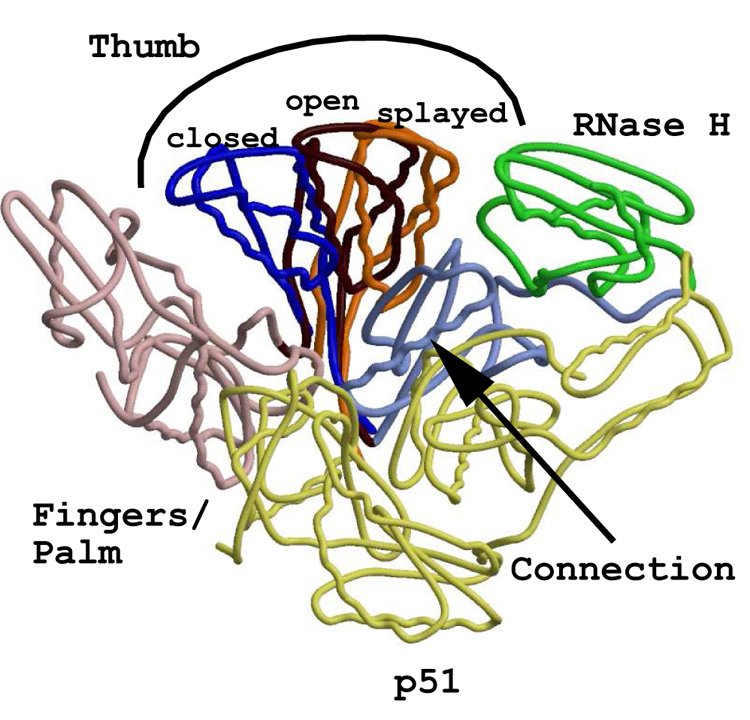
Superpositionings of three representative structures of HIV-1 RT, resulting in clearly distinguishable thumb positions. The p66 fingers/palm subdomain of the unliganded (lDLO) and an NNRTI-bound (lLWC) structure were superpositioned onto the p66 fingers/palm of the lHYS structure (HIV-1 RT complexed with RNA/DNA). For purposes of clarity, all domains and subdomains not involving the p66 thumb are those of the lHYS structure. The color scheme of the common motifs is as follows: p51 - light yellow; p66 fingers/palm - pink; p66 connection - light blue; RNase H - green. The “closed” (lDLO) thumb position is blue, the “open” (lHYS) thumb position is dark red, and the “splayed” (lLWC) thumb position is orange.
Three crystal structures of the unliganded HIV-1 RT heterodimer have been published (Esnouf et al., 1995; Hsiou et al., 1996; Rodgers et al., 1995), two of which represent examples of the “closed” structure, where the thumb subdomain virtually contacts the fingers, roughly forming the letter “O” (Hsiou et al., 1996; Rodgers et al., 1995). The third of these unliganded examples has the thumb in the “splayed” orientation, possibly as a consequence of the soaking out of the weakly bound NNRTI, HEPT (Esnouf et al., 1995). The structure assigned PDB code 1DLO will be used as the representative of the “closed” structure (Hsiou et al., 1996). The “open” thumb orientations have been observed for those structures in which HIV-1 RT is in complex with a nucleic acid duplex, wherein the opening between the fingers and the thumb forms a somewhat displaced “C”, essentially circumscribing a portal for the duplex to travel through the enzyme. Two nearly superimposable (root mean square deviation (rmsd) = 0.88 Å for residues 5–540 of p66), structurally related examples of the “open” structure are one with HIV-1 RT in a complex with dsDNA (PDB code 2HMI, (Ding et al., 1998)), and the other with an RNA/DNA hybrid (PDB code 1HYS, (Sarafianos et al., 2001)). Both of these crystal structures contain FAb28 for structural stabilization.
Interestingly, the structure of HIV-1 RT in a ternary complex with dsDNA and an incoming TTP has a rather unique “half-open” presentation, with part of the fingers subdomain closed down in interaction with the dNTP (Huang et al., 1998). The “splayed” thumb is seen in the crystal structures of HIV-1 RT in complex with NNRTIs, where the inhibitor binds in a unique hydrophobic pocket in the vicinity of the HIV-1 RT polymerase active site, virtually holding the thumb out in a V-like manner (Ren et al., 1995). Figure 2 shows the result of the superpositioning of the F/P subdomains (residues 4–243 in common) of the HIV-1 RT structures of 1DLO (the “closed” unliganded form) and the “splayed” NNRTI bound form (PDB code 1LWC, (Ren et al., 1995)) onto that of the “open” RNA/DNA bound form (1HYS) in order to underscore the importance of the flexibility of RTs via the movement of the thumb subdomain. Comparison of the HIV RT structures with that of the only known representative of a full-length MoMLV RT structure indicates that MoMLV RT clearly adopts the “open” form, consistent with an MoMLV RT-dsDNA bound complex.
6.2. Superpositioning analysis of MoMLV RT and HIV-1 RT
Several superpositionings between HIV-1 RT and MoMLV RT were performed to ascertain structural details and/or to optimize best fits for illustrations. Table 2 lists the structural motifs used in the superposition comparisons between MoMLV RT and HIV-1 RT. For purposes of optimal structural alignment, superpositionings involving only structurally conserved motifs were used, since inclusion of most of the loop regions contributed to poorer rmsd values as well as to far less meaningful visualizations of the commonalities between the structures. The HIV-1 RT structures possessing the most consistent and well-defined motifs for our study were 1HYS (Sarafianos et al., 2001) and 2HMI (Ding et al., 1998). The 2HMI structure was chosen since it is the complex most closely related to that of the MoMLV RT 1RW3 structure, having an 18/19-mer duplex DNA (encoding the DNA sequence of the PBS) poised at the polymerase active site of HIV-1 RT. The 1HYS structure was chosen since it contains an RNA/DNA hybrid representing the PPT. In addition, the RNase H domain of 1HYS is particularly well characterized; thus, its motifs were critical in RNase H comparisons as well. The superpositionings involving the RNases H were performed both in the context of the entire structure and as isolated domains (vide infra).
Table 2.
Common Regions for Superpositionings: MoMLV RT and HIV-1 RT
| Domain or Subdomain | MoMLV RT Residues | HIV-1 RT Residuesa | Secondary structure |
|---|---|---|---|
| Fingers/Palm | 68–84 | 28–43 | α-helix |
| 194–214 | 155–175 | α-helix | |
| 217–222 | 178–183 | β-strand | |
| 225–230 | 186–191 | β-strand | |
| 233–250 | 195–212 | α-helix | |
| 264–269 | 225–230 | β-strand | |
| 271–275 | 232–236 | β-strand | |
| Thumb | 295–308 | 255–268 | α-helix |
| 312–315 | 273–276 | β-strand | |
| 316–323 | 277–284 | α-helix | |
| 343–356 | 299–312 | α-helix | |
| Connection | 391–401 | 348–358 | β-strand |
| 411–427 | 366–382 | α-helix | |
| 439–449 | 395–405 | α-helix | |
| 452–457 | 408–413 | β-strand | |
| RNase H | 519–528 | 438–447 | β-strand |
| 533–540 | 452–459 | β-strand | |
| 547–552 | 463–468 | β-strand | |
| 559–573 | 475–489 | α-helix | |
| 577–582 | 492–497 | β-strand | |
| 584–592 | 499–507 | α-helix | |
| 615–626 | 516–527 | α-helix | |
| 629–634 | 530–535 | β-strand | |
| 648–657 | 544–553 | α-helix | |
Common regions listed here are from PDB code 1HYS.
Other HIV-1 RT structures might contain more gaps, thus fewer of the representatives listed were used in those comparisons.
The 2HMI structure has its RNase H extended to residue 558.
Table 4 is a compilation of a superpositioning analysis of MoMLV RT Model 1 onto the p66 of HIV-1 RT from the 1HYS structure, mathematically highlighting the different placements of the subdomains relative to each other. Similar values were obtained when the superpositionings were done with 2HMI and MoMLV RT Model 1 (data not shown). What is immediately obvious is that if the structural subdomains are superpositioned as separate entities, the rmsd values are quite good (~1–2 Å). The MoMLV RT F/P superpositioning onto 1HYS has an rmsd value of 1.32 Å (Table 4), which is statistically identical to that of HIV-1 RT p66 F/P of 1DLO onto 1HYS, and better than that of 1LWC onto 1HYS (Ren et al., 1995). However, when motifs from other subdomains are combined in the superpositionings between MoMLV RT and HIV-1 RT, the rmsd values increase, oftentimes dramatically. As an example, when the RNase H domain of Model 1 was superpositioned onto that of that of HIV-1 RT p66 RNase H, the rmsd value was quite respectable (2.45 Å for all motifs, see Table 4). However when the rotation/translation matrix was applied to the remainder of the molecule, virtually none of the rest of Model 1’s motifs coincided with their counterparts in HIV-1 RT, with the resulting structure’s thumb essentially pressing into the HIV-1 RT connection subdomain. This phenomenon is reflected mathematically in Table 4 when adding just one motif from the RNase H to the fingers/palm subdomain essentially tripled the rmsd value. The connection subdomain has the poorest single subdomain superpositioning, especially as the motifs nearing its C-terminus are added. This is due in part to the lack of structural characterization of MoMLV RT for the residues following β19 in the connection subdomain (residues 439–466). In this region HIV-1 RT has a defined α-helix and a β-strand (residues 395–418).
Table 4.
Superpositioning Results: MoMLV RT onto HIV-1 RT
| Secondary Structure | Rmsd (Å) |
|---|---|
| F/Pa: α-helices only | 1.26 |
| F/P: α-helices + 1st β-strand | 1.23 |
| F/P: α-helices + 1st & 2nd β-strands | 1.24 |
| F/P: α-helices + 1st, 2nd, & 3rd β-strands | 1.32 |
| F/P: 3 β-strands of P.A.S. only | 1.31 |
| F/P: All 4 β-strands | 1.32 |
| F/P: All motifs | 1.31 |
| F/P + 1st α-helix of Thumb | 2.30 |
| F/P 3 α-helices + 1st α-helix from Connection | 2.03 |
| F/P 3 α-helices + 1st α-helix + 1st β-strand from Connection | 2.44 |
| F/P All motifs + RNase H Model 1 | 6.72 |
| Thumb: α-helices only | 2.55 |
| Thumb: All motifs | 2.50 |
| Connection: α-helices only | 1.73 |
| Connection: All motifs | 3.48 |
| Connection + α-helices Thumb | 5.42 |
| RNase H: α-helices only | 1.15 |
| RNase H: β-strands only | 1.89 |
| RNase H: All Motifs | 2.45 |
F/P : fingers/palm
6.3. The movement in the connection/RNase H of MoMLV RT
Using the anisotropic network model in a normal mode analysis of MoMLV RT Model 1 in a comparison with the p66 of the 2HMI structure, it could be seen that long-range motions involving the hinge-bending of the connection subdomain relative to the RNase H domain strongly differ from those seen in HIV-1 (Atilgan et al., 2001). In the C-terminal portion of the connection, the residues 439–466 in MoMLV RT have motions strongly anticorrelated with the RNase H domain, whereas the residues in αM and β19 are strongly correlated with the RNase H (see Fig. 1). In HIV-1 RT, there is no real distinction in the motions of the corresponding regions of the connection subdomain; they are slightly anticorrelated throughout, with no sharp demarcations between the motifs (data not shown). The RNases H have sound structural superpositioning between MoMLV RT and HIV-1 RT, which is discussed in greater detail in section 9.
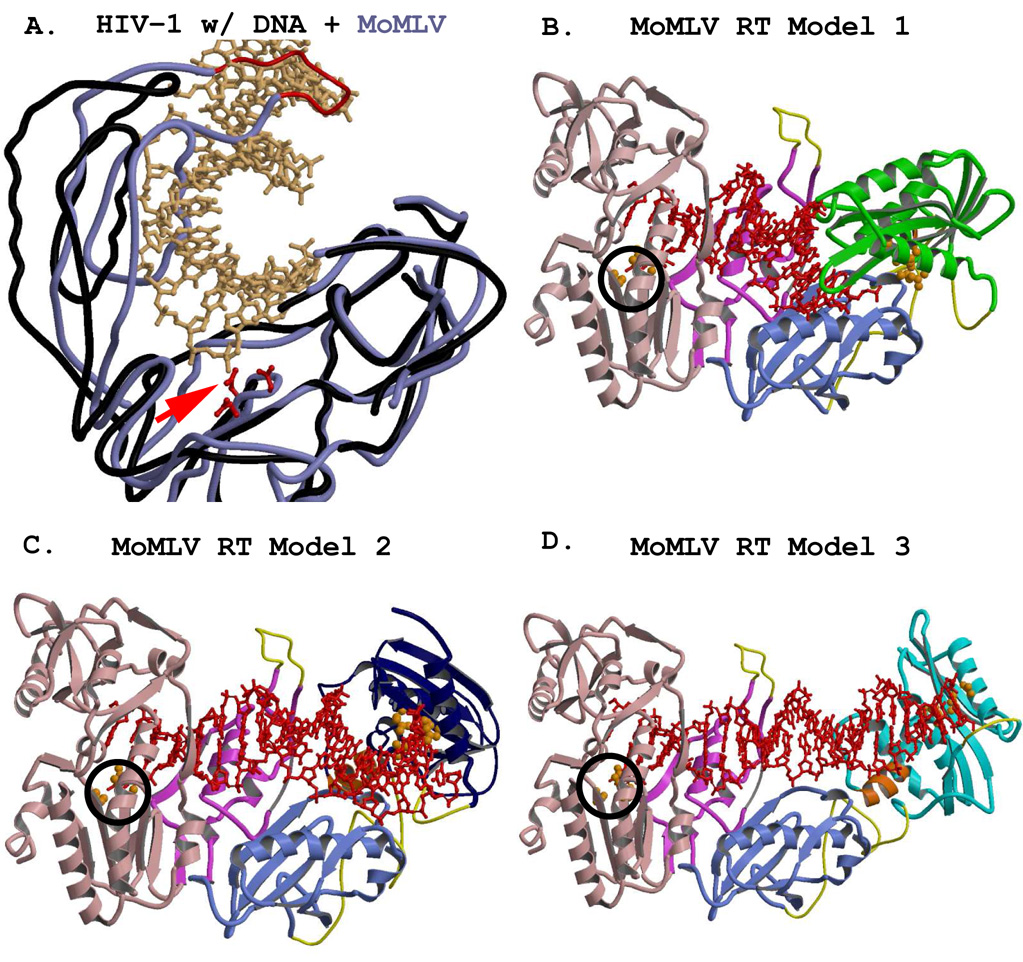
7. Full Length MoMLV RT Models
Single-molecule FRET studies and the use of locked nucleic acid analogs with HIV-1 RT suggest that different binding modes of template/primer can occur within the nucleic acid binding cleft of HIV-1 RT (Dash et al., 2004; Rothwell et al., 2003). It seems quite plausible, then, that the mobility of the RNase H domain of the monomeric MoMLV RT may span a broader distance range in MoMLV RT since there is no p51 analog present to take on the task of reorienting small substrates and/or of restricting the RNase H to a far tighter range of motion. Since the missing residues between the connection and RNase H of MoMLV RT have biochemical data attesting to their absolute necessity for the function of the enzyme (Puglia et al., 2006), it is possible that one of their essential functions could be to accommodate a wide range of movement of the RNase H domain. Therefore we propose two additional models of the full length MoMLV RT in light of the analysis performed from the purview of HIV-1 RT. All three MoMLV RT models retain the coordinates of the crystal structure of 1RW3 to residue 474; the placement of the RNase H domains is the only difference among them. In addition, the residues missing from the tip of the thumb in MoMLV RT 1RW3 were modeled (identically in all three models) such that they do not sterically clash with residues 365–368 in the connection subdomains of symmetry-related molecules in the crystal packing.
Model 1 corresponds to the 1RW3 structure with the RNase H domain placed into the experimental electron density (Das and Georgiadis, 2004) and is shown in Figure 3A. Although Model 1 accommodates the short substrate with which it was crystallized (see below), alternative models were tested which would accommodate longer substrates and allow a more general access to the RNase H active site.
Figure 3.
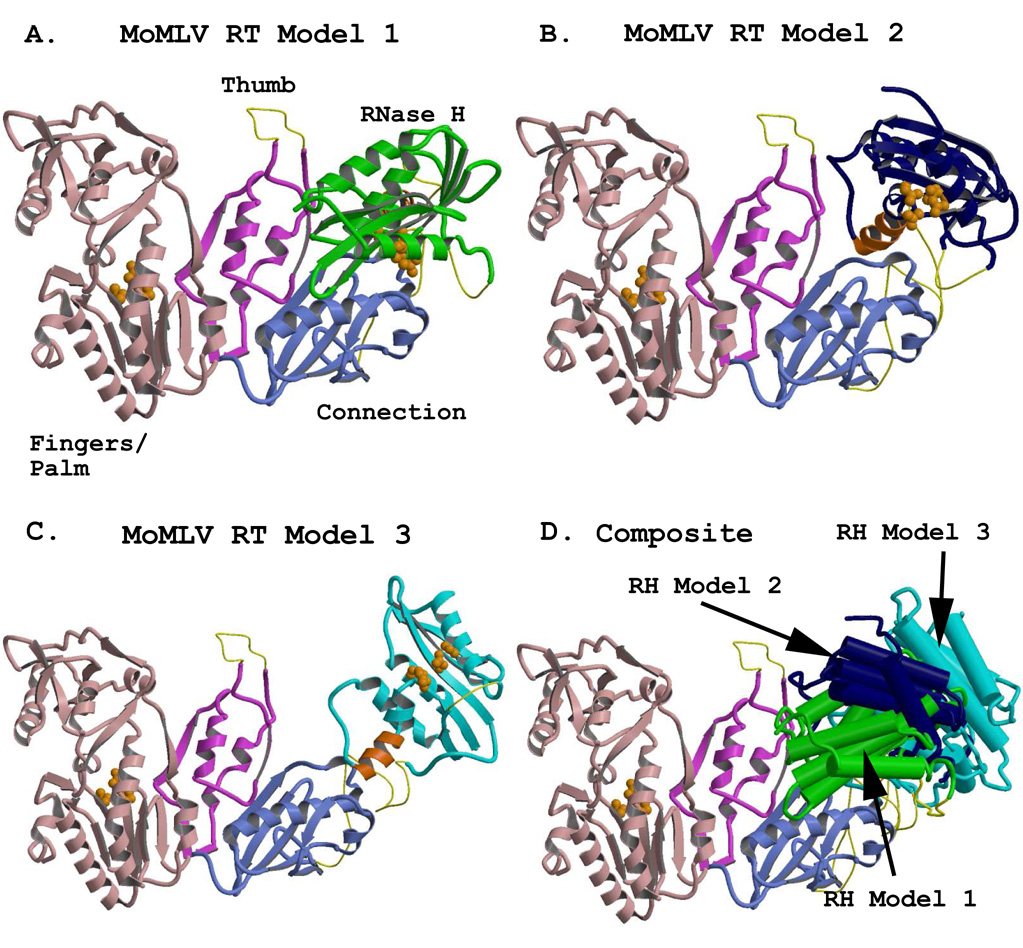
Representations of the full-length MoMLV RT structure, showing alternate positions of the RNase H domain. The models were generated using combinations of the crystal structure coordinates of PDB files 1RW3 and 2HB5. The fingers/palm, thumb, and connection subdomains are colored in light pink, fuscia, and light blue, respectively, and are rendered from the crystal structure coordinates of 1RW3. The RNase H domains were rendered from the coordinates of the crystal structure 2HB5. An added C-helix was modeled into the RNase H domains, and appears in orange. Regions missing from the reported crystal structures (see Table 3) are modeled with thin yellow connectors. Orange spheres represent the residues of the polymerase active site in the Fingers/Palm (F/P) and those of the DEDD active site in the RNases H. A) The full length MoMLV RT of Model 1 with its RNase H in green, occupying the position most closely fitted to the experimental electron density for the lRW3 crystal structure. B) MoMLV RT Model 2, with its RNase H in dark blue, rotated to a position +30° in y and +20° in x from its experimental locus, exposing both the C-helix and the RNase H active site. C) MoMLV RT Model 3, with its RNase H colored in cyan, superpositioned onto the placement of the RNase H as it occurs in the observed crystal structure of HIV-1 RT (PDB file 1HYS) and roated an extra 1.5° in the z direction. D) A superpositioning of the structures shown in A–C, with the structural elements of the RNases H rendered with cylinders for clarity. The colors of the RNases H correspond to those seen in the preceding panels.
To assist in the construction of the other two models, the C-helix/loop motif was added to the MoMLV RT RNaseH ΔC structure 2HB5. First, the C-helix motif and the two N- and C-terminally flanking residues of the E. coli RNase H (PDB code 2RN2, (Katayanagi et al., 1990)) structure were loaded into an O session (Jones et al., 1991) and rotated and translated close to the region into which they could reside in MoMLV RT RNase H. Next, the intact E. coli C-helix motif was “ligated” into the loop of the MoMLV RT RNase H ΔC. The amino acids were then changed to those encoded by MoMLV RT. The final structure was subsequently energy minimized using the AMBER suite of programs (Pearlman et al., 1995).
Model 2 (Fig. 3B) was created after having studied where the putative C-helix would reside in the RNase H domain of Model 1, as well as having investigated the scenarios involving the stretch of missing residues (475–502) unaccounted for in the combination of the two principal crystal structures yielding the full length MoMLV RT. In this model, a major design impetus was to maintain at least some of the RNase H molecule in reasonable coincidence with the location of the RNase H in Model 1. Thus, the RNase H was rotated roughly 20° in x and 30° in y around the center of mass of the RNase H of Model 1 (Model 2, Fig. 3B). This movement resulted in a large increase in the length of the cylindrical passageway needed for accommodation of longer intact nucleic acid duplexes (see below), and also allowed for a more accessible RNase H active site for longer substrates poised for polymerase-dependent RNase H activity. It also exposed the residues in the putative C-helix for unfettered interaction with the substrate. This C-helix motif, discussed in greater detail below, has been shown to be essential for proper recognition of the MoMLV RT PPT in vitro (Lim et al., 2002). Model 2 maintains the RNase H in approximately 60% of the area in which it resides in Model 1. In addition, Model 2 moves the start of the RNase H domain closer to the exit from the connection subdomain, imparting greater latitude for the type of structural motifs that may encompass the missing residues 475–502.
The RNase H domain was also repositioned in a third model (Model 3, Fig. 3C). The design of MoMLV RT Model 3 was a result of the study of the superpositionings of MoMLV RT with the various HIV-1 RT structures. Of the HIV-1 RT crystal structures investigated, the one with the RNase H in the most distant position from the polymerase active site was 1HYS, whose RNase H domain is 1.5 Å farther away than that of 2HMI. There also is a slightly larger opening (5.6 degrees) of the angle from the polymerase active site to the primer grip in the thumb to the RNase H active site in 1HYS. We thus considered the possibility that the position of the RNase H in MoMLV RT might closely emulate that of 1HYS at some stage during its reverse transcription cycle. A superpositioning of the F/P of 1RW3 onto that of the p66 of 1HYS indicated that the RNase H from 1HYS possibly could be accommodated as the RNase H domain in 1RW3. Thus, the common structural motifs of the isolated RHΔC structure (PDB code 2HB5) were superpositioned onto those of the RNase H of the superpositioned 1HYS structure, confirming that the location of the HIV-1 RT RNase H was compatible with the 1RW3 structure.
Pursuit of Model 3 would have ceased at this juncture, however, had it not been able to pack into the crystal lattice of 1RW3. No packing clashes or incompatibilities were observed for Model 3 in the lattice for MoMLV RT, an observation in and of itself suggesting that there indeed might be ample room for wide variations in the locus of the MoMLV RT RNase H. The putative C-helix/loop of MoMLV RT RNase H was subsequently modeled into its appropriate location. Model 3, therefore, is an effective hybrid of the MoMLV RT fingers/palm/thumb/connection with the RNase H as it occurs in HIV-1 RT (see Fig. 3C). It should be noted here that once the C-helix/loop was modeled into the MoMLV RT Model 3, some of those residues clashed with the connection subdomain. This was not surprising, since the superpositioning of the 1HYS RNase H onto the RHΔC, discussed in greater detail in section 9.4, shows that this modeled motif clashes with the connection subdomain of p66. As a result, a rotation of 1.5° in the +z-direction was applied to the MoMLV RT RNase H containing the C-helix/loop, to generate the final Model 3. This final Model 3 packed into the crystal lattice of 1RW3 with no clashes with any symmetry-related molecules. Figure 3C shows that the 475–502 “connector” in Model 3, represented by a completely randomized coiling, even if it were to contain a 20-residue α-helix, could easily accommodate the RNase H in a position highly reminiscent of HIV-1 RT itself. Figure 3D shows the superpositioning of the MoMLV RT fingers/palm/thumb and connection of Models 1, 2, and 3 with their resultant respective RNase H positions. The distance range spanned by the RNase H active sites of the models, as measured from the Mg2+ ion of a superpositioned RHΔC, is ~25Å. The distance spanned between the extreme edges of the RNases H in the full-length MoMLV RT models as measured from the E’ α-helix (Ala655) in Model 1 to the E’ α-helix (Arg657) in Model 3 is ~45Å.
8. MoMLV RT with Substrates
8.1. The bending of the DNA substrate in HIV-1 RT
The models of the full length MoMLV RT obviously should be considered in a similar context to the known models of HIV-1 RT, i.e., in complex with nucleic acid substrates. To that end, we investigated superpositionings of the MoMLV RT models with the HIV-1 RT 2HMI structure, which is that with a bound general sequence 18/19-mer dsDNA primer/template PBS emulate having the 3’OH of the primer strand properly positioned in the polymerase active site of HIV-1 RT. Figure 4A shows the p66 of the HIV-1 RT 2HMI structure with its bound DNA, as viewed from beyond the connection subdomain into the nucleic acid binding cleft. This view is identical in all of the panels of Figure 4. The DNA in the 2HMI structure has a helical bend of ~40°, occurring reasonably smoothly over a span of 4–5 base pairs starting at step #5 as it emanates from the polymerase active site and moves past the thumb. This bending is well known in polymerases, and involves the transition from A-form to B-form nucleic acid geometry, and is discussed extensively in several HIV-1 RT papers (Ding et al., 1998; Jacobo-Molina et al., 1993; Sarafianos et al., 2001).
Figure 4.
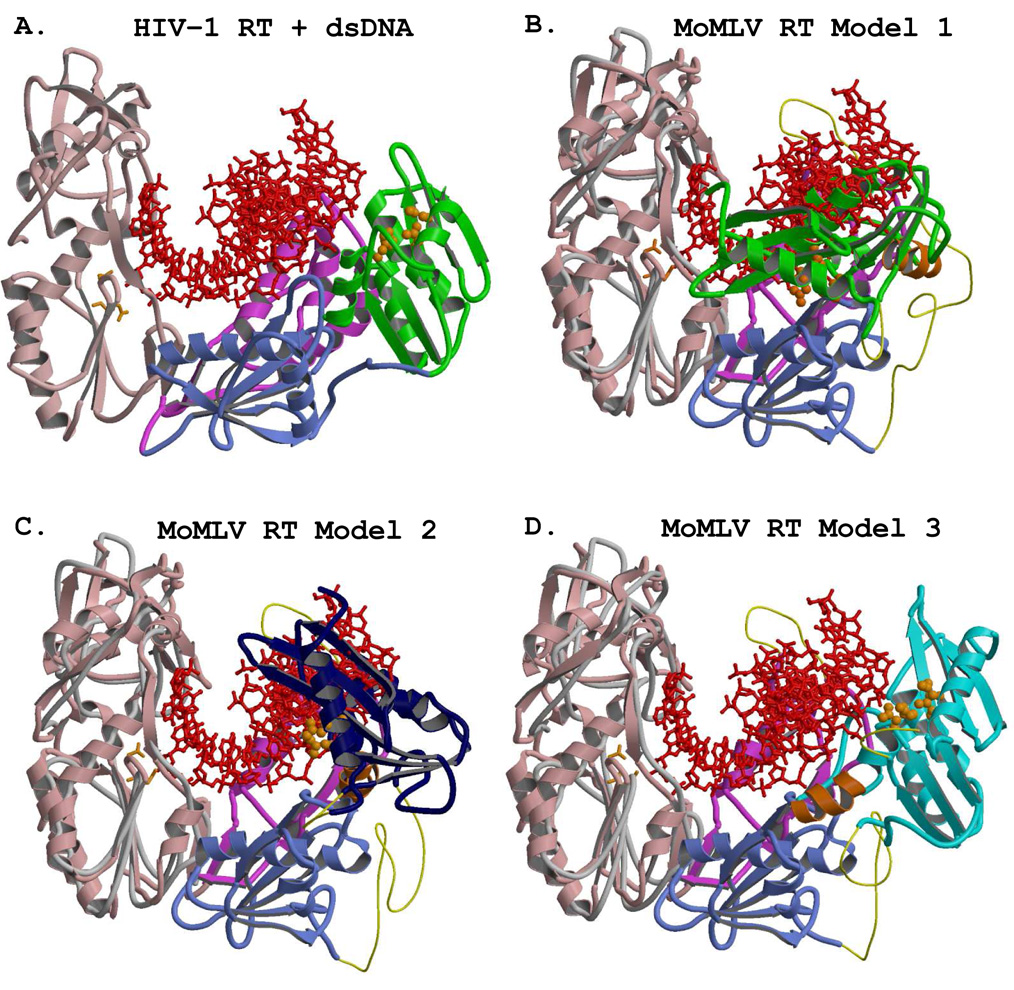
Superpositionings of the three MoMLV RT models onto the HIV-1 RT structure with its bound dsDNA substrate (PDB file 2HMI). The view, identical in all four panels, is from outside of the connection subdomain looking into the nucleic acid binding cleft. The position of the DNA of 2HMI remains constant for all four panels, and appears as a red ball-and-stick rendering. The active site residues of the respective RNases H are shown with orange spheres. The trace representing the fingers/palm of the HIV-1 RT, seen in panels B–D, is shown as a grey coil. A) The p66 subunit of the 2HMI structure with its bound 18/19-mer dsDNA. The fingers, palm, thumb, and connection are colored as in Fig. 2. The RNase H is green. The catalytic aspartates are those of the HIV-1 RT, and are shown as orange sticks. B) The superpositioning of the F/P of MoMLV RT Model 1 (with its RNase H in green) onto the F/P of p66 of 2HMI. The polymerase active site residues shown with orange sticks are those of MoMLV RT, as they are in panels C and D. C) The superpositioning of the same structural motifs for MoMLV RT Model 2, with its RNase H in dark blue. The wider channel created in Model 2 is apparent. D) The same superpositioning for MoMLV RT Model 3 onto HIV-1 RT, with its RNase H shown in cyan.
8.1.1. The MoMLV RT models compared with HIV-1 RT and its bound DNA
The conserved structural motifs of the fingers/palm subdomains of the models (see Table 2) were superpositioned onto the 2HMI HIV-1 RT structure, and the results of each superpositioning were examined. Since the N-termini of all of the MoMLV RT models are identical, the rmsd values were identical for all of these superpositionings (1.31 Å). It must be emphasized that for this particular analysis the nucleic acid of 2HMI was maintained in the position that it occupies in the HIV-1 RT structure. Figure 4B shows the result of the superpositioning of MoMLV RT Model 1 onto the 2HMI structure. It is clear that this model cannot accommodate the DNA in the position observed in HIV-1 RT, for the nucleic acid is essentially burrowing through the RNase H domain as positioned in Model 1. Even if the necessary further bending of the DNA were performed such that no clashes occur with the MoMLV RT thumb subdomain (vide infra), the 18/19-mer substrate still would not be easily accommodated in Model 1. Figure 4C shows the result for the superpositioning of MoMLV RT Model 2, also demonstrating that it cannot accommodate the nucleic acid duplex as it occurs in HIV-1 RT. The template-primers were positioned by triangulation with the catalytic residues in accordance with what is observed in the HIV-1 structures and further repositioned to avoid clashes with the MoMLV RT thumb subdomain. Thus, with appropriate bending of the DNA after proper positioning into the MoMLV RT polymerase active site, Model 2 could accommodate a rather long substrate, with ample opportunity for interaction with the C-helix and RNase H active site. MoMLV RT Model 3, with its RNase H placed into the HIV-1 RT biased position, is depicted in Figure 4D. This model also has the benefit in that the overall positioning of the RNase H domain accommodates the binding of large substrates. However, caution must be exercised in discussing any model not directly supported with structural evidence. The thumb position of MoMLV RT dictates that the trajectory of the nucleic acid would require a further movement of the RNase H of Model 3, approximately 5–10° more in z and ~ −5° in y (see Fig 4D), from the position that it occupies in HIV-1 RT to be poised for appropriate polymerase-dependent RNase H hydrolysis.
8.2. MoMLV RT models with DNA substrates
The motion of the thumb subdomain in HIV-1 RT is certainly not the only motion that occurs in this very flexible enzyme, and its variability of locus never occurs as an isolated event. It does, however, serve as a benchmark for the motions that do occur in the enzyme, since it is the subdomain observed to have the widest range of movement in HIV-1 RT (see Fig. 2). Since the full-length MoMLV RT was crystallized with DNA, its thumb subdomain clearly adopts the “open” confirmation in the structure, however it is turned more inward toward the connection/RNase H regions with its minor groove binding helix twisted slightly more away from the polymerase active site. Figure 5A shows the result of the superpositioning of all of the common structural motifs of the F/P of MoMLV RT onto those of the 2HMI structure of HIV-1 RT, with its bound DNA. The polymerase active site aspartates of MoMLV RT are shown as being incompatible with the 3’ OH of the DNA in the HIV-1 RT structure as positioned. Despite the near-coincident superpositioning of the polymerase active sites of the two enzymes (rmsd = 1.31 Å for the β-sheet comprising the respective polymerase active sites), it is clear that the thumb subdomain of MoMLV RT is directing its nucleic acid substrate in quite a different trajectory. Since it is known that the motions of the thumb in HIV-1 RT are correlated with those of the RNase H domain (Madrid et al., 2001; Temiz and Bahar, 2002), we considered models of the MoMLV RT with bound substrates in the context of the thumb/RNase H motions.
Figure 5.
The position of the MoMLV RT thumb subdomain directs the trajectory of the primer/template to a much different position than that of HIV-1 RT. A) Close up of the superpositioning of MoMLV RT onto HIV-1 RT with its dsDNA. The cutaway view is from the polymerase active site toward the RNase H domain. MoMLV RT is shown with a light blue coil, and the modeled thumb tip with a thinner red coil. The HIV-1 RT is in black. The polymerase active site residues Asp150, Asp224, and Asp225 of MoMLV RT are shown in red. The DNA is shown with beige ball-and-stick renderings. The required repositioning of the 3’ OH to the polymerase active site of MoMLV RT is indicated with a red arrow. In panels B – D the color scheme and the structural motifs are as they appear in Fig. 4. B) MoMLV RT Model 1, showing the repositioned 3’ OH (encircled in black) and further bending of the dsDNA of 2HMI, truncated to a 14-mer duplex, fitting properly into the experimental structure. C) MoMLV Model 2 with the entire 18/19-mer dsDNA of 2HMI, positioned appropriately into the MoMLV RT polymerase active site (circled in black). Less drastic bending of the DNA is required for extension into the RNase H domain and for favorable contacts to the C-helix (orange, behind the duplex) and RNase H active site. D) MoMLV RT Model 3 with the repositioned 3’ OH, accommodating the 29/31-mer RNA/DNA hybrid from the HIV-1 RT crystal structure lHYS.
8.2.1. The DNA bending in MoMLV RT Model 1
In Model 1, which is that of the experimental crystal structure, it was observed that the DNA from the HIV-1 RT structure would have to be translated approximately 1 base pair step (roughly 3 Å) for appropriate interaction of its 3’ OH with the MoMLV RT polymerase active site (see Figure 5A), then bent in excess of an additional 40° over 6–10 base pair steps after the fourth in order to avoid sterically clashing with the thumb and RNase H. In addition, it was observed that if the general sequence used from 2HMI were truncated to 14 base pairs, it would fit comfortably into the space allocated between the thumb and the RNase H of Model 1. Thus the DNA helix would need to be bent overall in excess of 80° in order to be accommodated by Model 1. Nucleic acid bending in excess of 100° has been observed in TATA-binding proteins (Masters et al., 2003), and CAP-DNA complexes (Napoli et al., 2006). In fact, in a survey of CAP-DNA complexes, the mean helical bend angle was found to be 80° +/− 12° (Kapanidis et al., 2001). The AT-rich duplex DNA used in the crystal structure of MoMLV RT has base pairs AT, GC, AT and TA as its first steps, preceded by a 5’ AT overhang. It has been reported that the duplex junction between the dG:dC/dA:dT steps in AT-rich nucleic acids can serve as a flexible hinge (Ng and Dickerson, 2002). Thus it is feasible that this feature of the nucleic acid used in the MoMLV RT structure possibly could have contributed to the greater bending of the substrate. Figure 5B shows Model 1 with its putative nucleic acid substrate appropriately positioned for MoMLV RT processing, in the most common view presented for reverse transcriptases. There is no clash with the thumb, and the bending of the nucleic acid (the 2HMI substrate translated, further bent and truncated) has not resulted in gross distortions of its structure. It is possible that this structure is an important example of an RT interacting with shorter substrates, and how the RNase H domain moves accordingly in a monomeric structure in order to establish the contacts necessary to initiate hydrolysis.
8.2.2. The DNA bending in MoMLV RT Model 2
The view of MoMLV RT Model 2 shown in Figure 4C depicts an open passageway through which a nucleic acid duplex could effortlessly pass. It is also quite clear that despite the open passageway created, the nucleic acid must still be reoriented and further bent in order to avoid the thumb and appropriately interact with the RNase H. Figure 5C shows MoMLV RT Model 2 with the repositioned 3’ OH and the full 18/19-mer duplex of 2HMI. The duplex was bent an additional 25° in order to avoid a clash with the thumb. The overall 65° helical bend is not uncommon for general sequence DNA interacting with a protein (Olson et al., 1998). As an aside, if the MoMLV RT thumb were in the position found in the HIV-1 RT “open” structures, then any additional bending of the DNA required for meaningful fitting into MoMLV RT Model 2 would be minimal, not exceeding 10° (data not shown). It can also be seen in Model 2 that the residues of the active site of the RNase H, the modeled C-helix (behind the DNA), and the modeled “His loop” are in favorable locations for enzymatic interaction. For Model 2, a general measure between the polymerase active site and the RNase H active site indicates that a nucleic acid substrate of approximately 17–18 base pairs could span that interval.
8.2.3. MoMLV RT Model 3 emulates HIV-1 RT p66
Figure 5D depicts the MoMLV RT Model 3 with the 29/31-mer primer/template RNA/DNA hybrid of the 1HYS structure, positioned into the MoMLV RT polymerase active site. There is also an observed helical bending of this substrate in the HIV-1 RT structure, similar in extent (~40°) and over similar base pair steps as that observed in the 2HMI structure (Sarafianos et al., 2001). Minimal manipulation of this substrate had to be performed for the proper repositioning into MoMLV RT Model 3, and an additional 8–10° bending of the helical axis ensured that the thumb subdomain did not impact its path. In addition, Figure 5D shows that MoMLV RT Model 3 also has the pertinent residues of the RNase H domain poised for appropriate interaction for polymerase-dependent RNase H activity. The isolated MoMLV RT RNase H domain, discussed below, examines these residues in greater detail. Base pair step distance measures indicate that a distance of approximately 20–21 base pairs is spanned between the polymerase and RNase H active sites in MoMLV RT Model 3.
8.3 Monomer vs. Heterodimer Structure/Function
Insights imparted by the MoMLV RT models as to its monomeric function in contrast to the functional heterodimeric HIV-1 RT are further elucidated by a chimeric RT construct wherein a 27 amino acid peptide from the MoMLV RT connection subdomain (residues 480–506) was inserted between residues 429 and 430 at the C-terminus of the HIV-1 RT connection subdomain (Pandey et al., 2001). This resulted in a functional monomeric enzyme, maintaining RNase H activity along with DNA synthesis on homopolymeric and heteropolymeric DNA templates and homopolymeric RNA templates. Interestingly, this chimera essentially contains the MoMLV connection tether region within 15 amino acids of the Trp repeat motif implicated in HIV-1 RT heterodimerization (Wapling et al., 2005). Insertion of the MoMLV connection tether region also blocked heterodimerization of the chimeric p66 domain with an exogenous p51 protein (Pandey et al., 2001). The latitude of motion afforded by the putative tether between MoMLV connection subdomain and RNase H domain is consistent with the partial interdependence of the two catalytic functions in the enzyme (Tanese and Goff, 1988). In contrast, structural constraints imposed by the heterodimer are reflected in the interdependence of the HIV-1 catalytic functions.
9. Comparisons of MoMLV RT and related RNases H
9.1. MoMLV RT RNase H and HIV-1 RNase H
The structural motifs of the RNases H share common α-helices and β-sheets such that superpositioning of these listed motifs yields rather low rmsd values (see Table 4). Two crystal structures of the isolated RNase H domain of HIV-1 RT have been published (PDB code 1HRH, (Davies et al., 1991); and PDB code 1RDH (Chattopadhyay et al., 1993)), as well as a solution (NMR) structure (Pari et al., 2003). The most structurally detailed of the isolated HIV-1 RT RNase H domains is that of PDB file 1HRH. The 1RDH HIV-1 RT RNase H structure is a low resolution structure, with only its Cα coordinates on deposit. In addition, the crystal structure of HIV-1 RT in complex with a polypurine tract RNA/DNA hybrid (1HYS) revealed important RNase H contacts with the substrate and thus is critical in the comparison with MoMLV RT (Sarafianos et al., 2001).
9.2. The structure of the isolated MoMLV RT RNase H
Figure 6A shows the ribbon rendering of the structure of the isolated MoMLV RT RNase HΔC (RHΔC; PDB code 2HB5, (Lim et al., 2006)). As mentioned earlier, the crystal structure does not possess the putative C-helix, or the stretch of residues in the “His loop” (see Fig. 1 and Fig. 6a). Both motifs were modeled into the MoMLV RT RNase H domain for purposes of discussion but are not shown in Figure 6A, which depicts the actual structure crystallized. The RHΔC structure contains one Mg2+ ion chelated by the three aspartic acid residues and the one glutamic acid residue of the D-E-D-D motif. Two of the residues of this RNase H active site (Glu562 and Asp653) motif were found to adopt two distinct conformations in the crystal structure. Figure 6A shows Glu562 and Asp653 in the conformation designated “A” in the PDB file. Other residues observed to have two distinct side-chain conformations were Gln533, Arg534, Glu545 Gln559, Lys571, Lys576, Glu610, Ser631 and Asn649 (see Fig. 1), most of which reside in the shallow depression immediately preceding the RNase H active site. The positively-charged residues listed above have been identified in structural and biochemical studies as being associated with substrate binding (Blain and Goff, 1993; Blain and Goff, 1995; Kanaya et al., 1991a; Telesnitsky and Goff, 1993), thus their dual conformations in the RHΔC structure may have some biological relevance, and this may also be the case regarding the two conformations of the active site residues.
Figure 6.
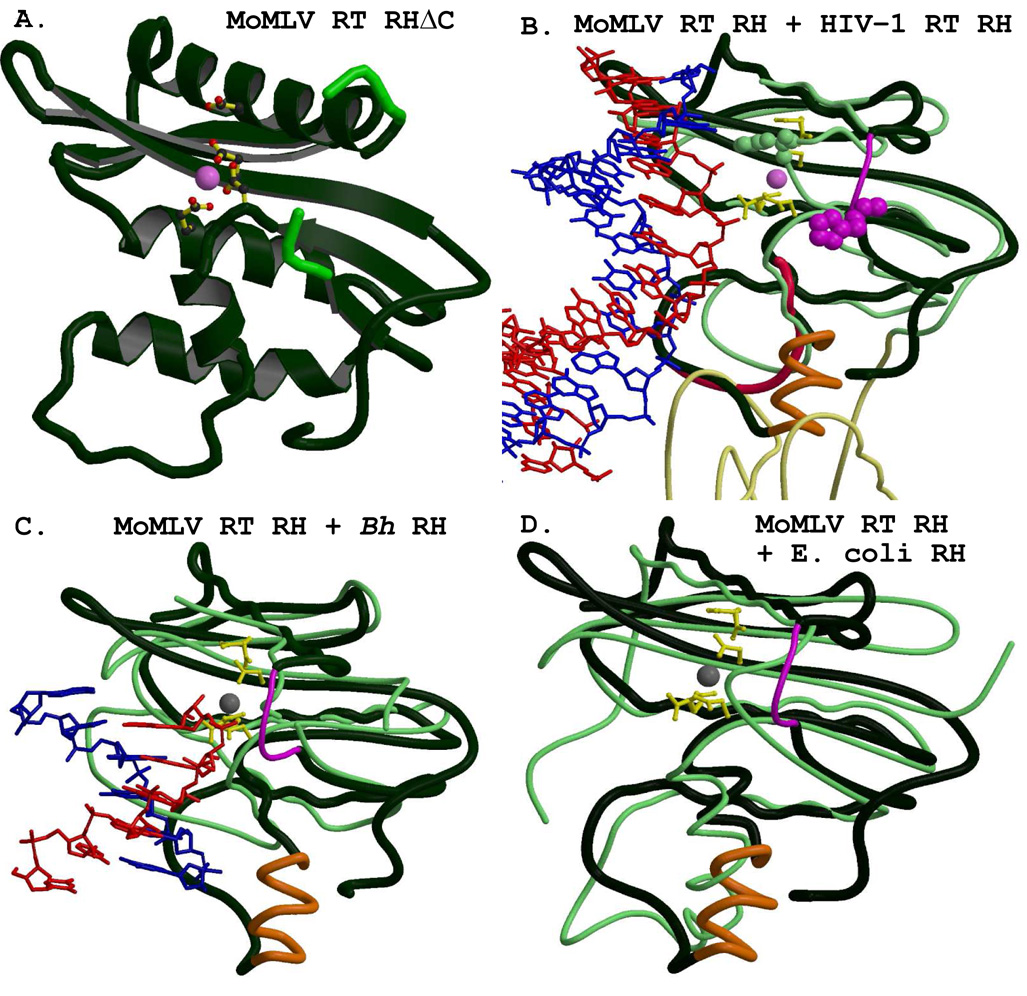
The isolated MoMLV RT RNase H structure has great structural similarity with HIV-1 RT and other RNases H. A) The isolated MoMLV RT RNase HΔC structure (PBD code 2HB5) is shown in dark green, consistent for all of the panels, with its bound Mg2+ shown as a pink sphere. The catalytic residues Asp524, Glu562, Asp583, and Asp653 (with Glu562 and Asp653 shown in their “A” conformations) are rendered as colored ball-and-stick figures. Residues His634, Cys635, Gly641, and His642 are shown in lighter green to emphasize the break in the chain for the missing residues 636 – 640. The panels retain a similar viewpoint throughout. B) The superpositioning of the RNase H of the 1HYS structure (rendered with thin light green coils) with its bound RNA/DNA hybrid onto that of MoMLV RT RNase HΔC. The catalytic residues of MoMLV RT RNase H are shown, with both conformations of Glu562 and Asp653, as yellow ball-and-stick figures, as they are in the following panels. The bound Mg2+ is shown as a pink sphere. A thinner magenta coil, appearing just to the right of the Mg2+, represents the modeled residues of the missing “His loop” for this and the following panels. The actual trace of the MoMLV RT RNase HΔC is shown with a crimson coil, and the modeled C-helix is rendered in orange. The HIV-1 RT p66 connection subdomain, shown at the bottom of the figure, appears in thinner light yellow coils. The RNA template is colored red, and the DNA primer is colored blue. His539 of HIV-1 RT is shown with light green spheres. The sequence-aligned analog of MoMLV RT, His638, not characterized in the crystal structure, is shown with magenta spheres to emphasize the differences in the respective loci of the His-loops in the two enzymes. C) The superpositioning of the RNase H of B. halodurans (Bh, PDB code 2G8H, in thin light green coils) with its bound RNA/DNA (red/blue) onto MoMLV RT RNase HΔC. The Mg2+ appears as a grey sphere. D) A superpositioning of the RNase H from E. coli (PDB code 2RN2, in thin green coils) onto the MoMLV RT RNase HΔC. The motifs of MoMLV RT are the same as seen in 6c above. Note how the positioning of the modeled C-helix corresponds to the actual structure of the E. coli RNase H.
9.3. Magnesium in the RNase H structures
Structural work by Nowotny et al. discusses the various retroviral and retrotransposon RNases H as having a 1- or 2-Mg2+ ion dependent activity (Nowotny et al., 2005; Nowotny and Yang, 2006). They contend that despite several crystal structures of RNases H containing only one magnesium ion bound in the RNase H active site, they are 2-Mg2+- binding species, and the presence of both Mg2+ likely depends upon the presence of a nucleic acid substrate. The MoMLV RT RNase HΔC crystal structure contains one Mg2+ ion in the RNase H active site. The two distinct conformations for Glu562 and Asp653 may have some biological relevance, since their interactions with the Mg2+ in the structure are not equivalent. The Mg2+ crystallized in the MoMLV RT structure corresponds to the non-activating transition-state stabilizing magnesium (metal ion “B” as described in (Nowotny and Yang, 2006)) of a 2-Mg2+ ion dependent RNase H. The Mg2+ ion observed in the MoMLV RT RNase HΔC structure also forms bonds to two water oxygens, critical in the process of RNA hydrolysis (Kanaya et al., 1996; Nowotny et al., 2005).
9.4 Superpositionings of MoMLV RT RNase H and HIV-1 RT RNase H
The isolated HIV-1 RT RNase H structure 1HRH crystallized with two molecules in its asymmetric unit, and neither molecule (A nor B) is structured within the “His Loop” (see Fig. 1). The molecule designated “A” was investigated for superpositioning studies with MoMLV RT since its C-terminal helix extends to residue Arg537, whereas that of molecule “B” terminates at Gly555. A superpositioning of the common structural motifs of 1HRH molecule A with those of 2HB5 gave rmsd values of 1.5 Å and 2.7 Å for α-helices only and all motifs, respectively. These values are slightly higher than those observed for the same RNase H motifs from the 1HYS structure, so all common motifs of the RNase H domain of the 1HYS structure (with its bound RNA/DNA hybrid) were superpositioned with the MoMLV RT RNase HΔC. It should be mentioned here that the 29/31-mer hybrid of 1HYS contains the sequence of the HIV-1 RT PPT, which has weakly paired, unpaired and mispaired bases in its structure starting at the RNase H primer grip and proceeding to the RNase H active site. The presence of structural deformations seen in the uncomplexed PPT indicates that this may not be a property applicable to general sequences (Kvaratskhelia et al., 2002). Furthermore, the RNase H active site in IHYS is ~3Å from the predicted scissile phosphate within the RNA of the PPT (Sarafianos et al., 2002). Figure 6B shows the result of the superpositioning of the common RNase H structural motifs listed in Table 2. The figure contains both the actual RHΔC (the difference is highlighted by a red coil) and the modeled additional C-helix and “His loop” for discussion purposes.
Figure 6B shows two regions where the structure of MoMLV RT RNase H differs greatly from that of the HIV-1 RT RNase H. First, in either the MoMLV RT RHΔC or the structure containing the modeled C-helix/loop motif, the “His loop” is shown as a thin magenta coil between β’5 and α-helix E’ (see also Figure 1) of the MoMLV RT. This loop was modeled in attempt to follow the trajectory indicated by Cys635 and His642 in the PDB file, and is not meant to reflect any experimental data. In this modeling, the His loops of the two structures do not appear to be able to easily coincide. Given the trajectory of the Cys635 and His642, however, it seems that even if the actual loop were positioned to better emulate that of HIV-1 RT, it certainly would not coincide with it. Two of the five missing residues from the His loop of RHΔC are conserved: Pro636 and His638 in MoMLV RT, corresponding to Pro537 and His539 of HIV-1 RT. In the HIV-1 RT structure shown in Figure 6B, the bend of the thin light green coil immediately to the right of the Mg2+ shows, via filled-spheres, the location of the conserved His539 of its His loop. The figure also indicates that for proper positioning of a putative substrate by this His loop and C-helix for interaction with the MoMLV RT RNase H, the substrate would require a rotation of ~10°. Initially, when the putative C-helix/loop motif was added to the RNase H domain in the HIV-1 RT emulate Model 3, it clashed with the connection subdomain. This point is illustrated in Figure 6B, using the isolated RNase H domain of MoMLV RT superpositioned with the actual 1HYS HIV-1 RT structure. Two loops from the HIV-1 RT p66 connection (light yellow coils), representing residues 359–362 (part of the HIV-1 RT RNase H primer grip) and 402–406, overlap the residues of the C-helix/loop region modeled into the isolated MoMLV RT RNase H. A similar clash occurred in the initial building of the MoMLV RT Model 3, which was corrected by a 1.5° rotation of RNase H in the model.
9.5. Superpositionings of MoMLV RT RNase H and other RNases H
Recent structural advances of bacterial RNases H warrant comparison with the MoMLV RT RNase H domain. The structure of an inactive mutant (Asp192Asn) B. halodurans RNase H complexed to an RNA/DNA substrate has been obtained (PDB code 2G8H; (Nowotny and Yang, 2006)) and the common structural motifs were superpositioned onto the MoMLV RT RHΔC. Figure 6C shows this superpositioning (rmsd = 3.23 Å for all common motifs). The non-activating transition-state mediating magnesium in B. halodurans RNase H (Nowotny et al., 2005; Nowotny and Yang, 2006) corresponded with the Mg2+ of MoMLV RHΔC to 1.5 Å (shown as a grey sphere). The loop motif (in thin green coils, corresponding to residues 142–150) used by B. halodurans for substrate positioning is clearly not in common with MoMLV RT, despite the close coincidence of their respective Mg2+. Thus it seems that this region, used for positioning the substrate in the respective RNases H, is clearly the most variable between the two enzymes.
The first crystal structure of an isolated RNase H domain was that of E. coli RNase H (PDB code 2RN2, (Katayanagi et al., 1990)). This RNase H structure is the one with which MoMLV RT shares a putative α-helix, the C-helix. Figure 6D shows the superpositioning of the common structural motifs of the 2RN2 structure (in light green coils) onto those of RHΔC (rmsd = 2.56 Å). The His loop motif of E. coli is well characterized in the structure, and in fact nearly coincides in shape and trajectory with that of HIV-1 RT RNase H (data not shown), as can be seen in comparison to Figure 6B. The approximate coincidence of the two C-helices in the 2RN2 versus RHΔC structures indicates that these two enzymes may position their respective substrates in much the same manner. Interestingly, introduction of the E. coli C-helix into an isolated HIV-1 RT RNase H domain restored its RNase H activity (Keck and Marqusee, 1995).
Conclusion
The similarities between the monomeric MoMLV RT and heterodimeric HIV-1 RT are intriguing, given the large differences between the protein sequences and the number of residues required to perform the same functions. Structural comparisons between the individual subdomains of MoMLV and those of the HIV-1 RT p66 subunit highlight the remarkable fold similarities between the two enzymes. The largest differences lay in the relative positioning of the subdomains with respect to each other and to the trajectory of the putative nucleic acid substrates.
A major structural difference between MoMLV RT and HIV-1 RT resides in the connection subdomain, where the C-terminal portion of MoMLV RT has less well defined secondary structure than does HIV-1 RT. The 32 amino acid stretch of residues between the connection and RNase H of MoMLV RT has no analog in HIV-1 RT p66, suggesting that it may in fact serve as a tether supporting a wider range of motion of the RNase H. Molecular modeling of the MoMLV RT structure reported in 1RW3 with short DNA substrates indicates that the position of the RNase H domain is consistent with the presence of a 14-mer substrate. Two alternative models (Models 2 and 3, Figs. 3B, 3C), consistent with the crystal packing of the full-length MoMLV RT structure, are proposed in which the RNase H domain has been transposed to allow for binding of longer nucleic acid substrates, accommodate the α-C helix, and expose the RNase H catalytic triad. Although the size of the RNA/DNA hybrid that can span the distance between the DNA polymerase and RNase H active sites differ in Models 2 and 3 (17–18 vs. 20–21 bp, respectively), both models are consistent with biochemical data for polymerase-dependent RNase H cleavages (Schultz et al., 2006). The larger range of motion of the RNase H of MoMLV RT versus HIV-1 RT seems to be required to accommodate the full spectrum of its activity. This quite possibly may be the essence of a monomeric versus a heterodimeric enzyme performing the same function.
Table 3.
Amino acid residues missing in the available crystal structures of MoML V RT a
| Fingers/Palm: | 1–23 |
| Thumb (tip): | 327–334 |
| Connection/RNaseH: | 475–502 |
| RNase H (C-helix/loop): | 593–603 |
| RNase H (“His loop”): | 636–640 |
| RNase H (C-terminus): | 669–671 |
PDB code 1RW3 (Das and Georgiadis, 2004); PDB code 2HB5 (Lim et al., 2006)
Acknowledgements
This work was supported by NIH grant RO1 GM070837 issued to MR. We thank William Schneider for his critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Figures Figure 2–Figure 6 were prepared by using O (Jones et al., 1991), MOLSCRIPT (Kraulis, 1991), and RASTER3D (Merritt and Bacon, 1997).
References
- Abbink T, Berkhout B. HIV-1 reverse transcription initiation: a potential target for novel antivirals? Virus Res. 2008 doi: 10.1016/j.virusres.2007.12.009. in press. [DOI] [PubMed] [Google Scholar]
- Atilgan A, Durell S, Jernigan R, Demirel M, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K, Beard WA, Casas-Finet JR, Kim HR, Darden TA, Wilson SH, Kunkel TA. Reduced frameshift fidelity and processivity of HIV-1 reverse transcriptase mutants containing alanine substitutions in helix H of the thumb subdomain. J. Biol. Chem. 1995;270:19516–19523. doi: 10.1074/jbc.270.33.19516. [DOI] [PubMed] [Google Scholar]
- Blain SW, Goff SP. Nuclease activities of Moloney murine leukemia virus reverse transcriptase. J. Biol. Chem. 1993;268:23585–23592. [PubMed] [Google Scholar]
- Blain SW, Goff SP. Effects on DNA synthesis and translocation caused by mutations in the RNase H domain of Moloney murine leukemia virus reverse transcriptase. J. Virol. 1995;69:4440–4452. doi: 10.1128/jvi.69.7.4440-4452.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PL, Gao HQ, Frank P, Clark PK, Hughes SH. The basic loop of the RNase H domain of MLV RT is important both for RNase H and for polymerase activity. Virology. 2001;282:206–213. doi: 10.1006/viro.2000.0827. [DOI] [PubMed] [Google Scholar]
- Boyer PL, Julias JG, Ambrose Z, Siddiqui MA, Marquez VE, Hughes SH. The nucleoside analogs 4′C-methyl thymidine and 4′C-ethyl thymidine block DNA synthesis by wild-type HIV-1 RT and excision proficient NRTI resistant RT variants. J Mol Biol. 2007;371:873–882. doi: 10.1016/j.jmb.2007.05.043. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay D, Finzel BC, Munson SH, Evans DB, Sharma SK, Strakalaitis NA, Brunner DP, Eckenrode FM, Dauter Z, Betzel C, Einspahr HM. Crystallographic analyses of an active HIV-ribonuclease H domain show structural features that distinguish it from the inactive form. Acta Crystallog., Sect. D. 1993;49:423–427. doi: 10.1107/S0907444993002409. [DOI] [PubMed] [Google Scholar]
- Coffin JM, Hughes SH, Varmus HE. Reverse transcriptase and the generation of retroviral DNA. Retroviruses Chapter 1. New York: Cold Spring Harbor Laboratory Press; 1997. pp. 121–160. [PubMed] [Google Scholar]
- Coté ML, Georgiadis MM. Crystal structure of a pseudo-16-mer DNA with stacked guanines and two G-A mispairs complexed with the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 9):1238–1250. doi: 10.1107/s090744490100943x. [DOI] [PubMed] [Google Scholar]
- Coté ML, Yohannan S, Georgiadis MM. Use of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase to facilitate crystallization and analysis of a pesudo-16-mer DNA molecule containing G-A mispairs. Acta Cryst. 2000;D56:1120–1131. doi: 10.1107/s0907444900008246. [DOI] [PubMed] [Google Scholar]
- Das D, Georgiadis M. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure (Camb) 2004;12:819–829. doi: 10.1016/j.str.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Das D, Georgiadis MM. A directed approach to improving the solubility of Moloney murine leukemia virus reverse transcriptase. Protein Sci. 2001;10:1936–1941. doi: 10.1110/ps.16301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash C, Yi-Brunozzi H-Y, Le Grice SFJ. Two modes of HIV-1 polypurine tract cleavage are affected by introducing locked nucleic acid analogs into the (−) DNA template. J. Biol. Chem. 2004;279:37095–37102. doi: 10.1074/jbc.M403306200. [DOI] [PubMed] [Google Scholar]
- Davies JF, 2nd, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- Ding J, Das K, Hsiou Y, Sarafianos SG, A D, C J, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- Ding J, Das K, Moerells H, Koymans L, Andries K, Janseen PA, Hughes SH, Arnold E. Structure of HIV-1 RT/TIBO R86183 reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995a;2:407–415. doi: 10.1038/nsb0595-407. [DOI] [PubMed] [Google Scholar]
- Ding J, Das K, Tantillo C, Zhang W, Clark AD, Jr, S J, Lu X, Hsiou Y, Jacobo-Molina A, Andries K, Pawels R, Moereels H, Koymans L, Janssen PAJ, Smith RH., Jr Structure of HIV-1 reverse transcriptase in a complex with the nonnucleoside inhibitor a-APA R95845 at 2.8A resolution. Structure. 1995b;3:365–379. doi: 10.1016/s0969-2126(01)00168-x. [DOI] [PubMed] [Google Scholar]
- Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- Furfine ES, Reardon JE. Reverse transcriptase-RNase H from the human immunodeficiency virus. J. Biol. Chem. 1991;266:406–412. [PubMed] [Google Scholar]
- Georgiadis MM, Jessen SM, Ogata CM, Telesnitsky A, Goff SP, Hendrickson WA. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure. 1995;3:879–892. doi: 10.1016/S0969-2126(01)00223-4. [DOI] [PubMed] [Google Scholar]
- Gille C, Frömmel C. STRAP: editor for STRuctural Alignments of Proteins. Bioinformatics. 2001;17:377–378. doi: 10.1093/bioinformatics/17.4.377. [DOI] [PubMed] [Google Scholar]
- Gu J, Villanueva R, Snyder C, Roth M, Georgiadis M. Substitution of Asp114 or Arg116 in the fingers domain of Moloney murine leukemia virus reverse transcriptase affects interactions with the template-primer resulting in decreased processivity. J Mol. Biol. 2001;305:341–359. doi: 10.1006/jmbi.2000.4281. [DOI] [PubMed] [Google Scholar]
- Hsiou Y, Ding J, Das K, Clark ADJ, Lu X, Tantillo C, Williams R, Kramer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Okumura M, Katayanagi K, Kimura S, Kanaya S, Nakamura H, Morikawa K. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 A resolution. J Mol Biol. 1993;23:529–542. doi: 10.1006/jmbi.1993.1169. [DOI] [PubMed] [Google Scholar]
- Jacobo-Molina A, Ding J, Nanni RG, Arthur D, Clark J, Lu X, Tantilla C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager J, Smerdon SJ, Wang J, Boisvert DC, Steitz TA. Comparison of three different crystal forms shows HIV-1 reverse transcriptase displays an internal swivel motion. Structure. 1994;2:869–876. doi: 10.1016/s0969-2126(94)00087-5. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron denisty maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kanaya S, Katsuda C, Kimura S, Nakai T, Kitakuni E, Nakamura H, Katayanagi K, Morikawa K, Ikehara M. Stabilization of Escherichia coli ribonuclease H by introduction of an artificial disulfide bond. J. Biol. Chem. 1991a;266:6038–6044. [PubMed] [Google Scholar]
- Kanaya S, Katsuda-Nakai C, Ikehara M. Importance of the positive charge in Escherichia coli ribonuclease HI for the effective binding of the substrate. J. Biol. Chem. 1991b;266:11621–11627. [PubMed] [Google Scholar]
- Kanaya S, Oobatake M, Liu Y. Thermal stability of Escherichia coli ribonuclease HI and its active site mutants in the presence and absence of the Mg2+ ion. Proposal of a novel catalytic role for Glu48. J Biol Chem. 1996;271:32729–32736. doi: 10.1074/jbc.271.51.32729. [DOI] [PubMed] [Google Scholar]
- Kapanidis AN, Ebright YW, Ludescher RD, Chan S, Ebright RH. Mean DNA bend angle and distribution of DNA bend angles in the CAP-DNA complex in solution. J. Mol Biol. 2001;312:453–468. doi: 10.1006/jmbi.2001.4976. [DOI] [PubMed] [Google Scholar]
- Katayanagi K, Miyagawa M, Matasushima M, Ishikawa M, Kanyana S, Nakamura H, Ikehara M, Matsuzaki T, Morikawa K. Structural details of ribonuclease H from Escherichia coli as refined to an atomic resolution. J. Mol. Biol. 1992;223:1029–1052. doi: 10.1016/0022-2836(92)90260-q. [DOI] [PubMed] [Google Scholar]
- Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- Keck JL, Marqusee S. Substitution of a highly basic helix/loop sequence into the RNase H domain of human immunodeficiency virus reverse transcriptase restores its Mn2+ - dependent activity. Proc. Natl. Acad. Sci. USA. 1995;92:2740–2744. doi: 10.1073/pnas.92.7.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- Kvaratskhelia M, Budihas S, Grice SL. Pre-existing distortions in nucleic acid structure aid polypurine tract selection by HIV-1 reverse transcriptase. J. Biol. Chem. 2002;277:16689–16696. doi: 10.1074/jbc.M109914200. [DOI] [PubMed] [Google Scholar]
- Leis J, Aiyar A, Cobrinik D. Regulation of initiation of reverse transcription of retroviruses. In: Skalka AM, Goff SP, editors. Reverse Transcriptase. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1993. pp. 33–47. [Google Scholar]
- Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP. Crystal structure of the Moloney murine leukemia virus RNase H domain. J. Virol. 2006;80:8379–8389. doi: 10.1128/JVI.00750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Orlova M, Goff SP. Mutations of the RNase H C helix of the Moloney murine leukemia virus reverse transcriptase reveal defects in polypurine tract recognition. J. Virol. 2002;76:8360–8373. doi: 10.1128/JVI.76.16.8360-8373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid M, Lukin JA, Madura JD, Ding J, Arnold E. Molecular dynamics of HIV-1 reverse transcriptase indicates increased flexibility upon DNA binding. Proteins: Structure, Function, and Genetics. 2001;45:176–182. doi: 10.1002/prot.1137. [DOI] [PubMed] [Google Scholar]
- Masters KM, Parkhurst KM, Daugherty MA, Parkhurst LJ. Native human TATA-binding protein simultaneously binds and bends promoter DNA without a slow isomerization step or TFIIB requirement. J Biol Chem. 2003;278:31685–31690. doi: 10.1074/jbc.M305201200. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ. Raster3d: Photorealistic molecular graphics. Methods Enzymol. 1997;227:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Mulky A, Sarafianos SG, Arnold E, Wu X, Kappes JC. Subunit-specific analysis of the human immunodeficiency virus type 1 reverse transcriptase in vivo. J. Virol. 2004;78:7089–7096. doi: 10.1128/JVI.78.13.7089-7096.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmudin S, Coté ML, Sun D, Yohannan S, Montano SP, Gu J, Georgiadis MM. Crystal structures of an N-terminal fragment from Moloney murine leukemia virus reverse transcriptase complexed with nucleic acid: Functional implications for template-primer binding to the fingers domain. J. Mol. Biol. 2000;296:613–632. doi: 10.1006/jmbi.1999.3477. [DOI] [PubMed] [Google Scholar]
- Napoli AA, Lawson CL, Ebright RH, Berman HM. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: recognition of pyrimidine-purine and purine-purine steps. J. Mol Biol. 2006;357:173–183. doi: 10.1016/j.jmb.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HL, Dickerson RE. Mediation of the A/B-DNA helix transition by G-tracts in the crystal structure of duplex CATGGGCCCATG. Nucleic Acids Res. 2002;30:4061–4067. doi: 10.1093/nar/gkf515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: Substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollis DL, Kline C, Steitz TA. Domain of E. coli DNA polymerase I showing sequence homology to T7 DNA polymerase. Nature. 1985;313:818–819. doi: 10.1038/313818a0. [DOI] [PubMed] [Google Scholar]
- Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P, Kaushik N, Talele T, Yadav P, Pandey V. Insertion of a peptide from MuLV RT into the connection subdomain of HIV-1 RT results in a functionally active chimeric enzyme in monomeric conformation. Mol Cell Biochem. 2001;225:135–144. doi: 10.1023/a:1012278308154. [DOI] [PubMed] [Google Scholar]
- Pari K, Mueller GA, DeRose EF, Kirby TW, London RE. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry. 2003;42:639–650. doi: 10.1021/bi0204894. [DOI] [PubMed] [Google Scholar]
- Pearlman DA, Case DA, Caldwell JW, Ross WR, Cheatham TE, III, DeBolt S, Ferguson D, Seibel G, Kollman P, Amber P. AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structure and energies of molecules. Comp. Phys. Commun. 1995;91:1–41. [Google Scholar]
- Powers R, Clore GM, Bax A, Garrett DS, Stahl SJ, Wingfield PT, Gronenborn AM. Secondary structure of the ribonuclease H domain of the human immunodeficiency virus reverse transcriptase in solution using three-dimensional double and triple resonance heteronuclear magnetic resonance spectroscopy. J. Mol. Biol. 1991;221:1081–1090. doi: 10.1016/0022-2836(91)90920-2. [DOI] [PubMed] [Google Scholar]
- Puglia J, Wang T, Smith-Snyder C, Coté M, Scher M, Pelletier J, John S, Jonsson C, Roth M. Revealing domain structure through linker-scanning analysis of the murine leukemia virus (MuLV) RNase H and MuLV and human immunodeficiency virus type 1 Integrase proteins. J. Virol. 2006;80:9497–9510. doi: 10.1128/JVI.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Esnouf R, Garman E, Somers D, Ross C, Kiraby I, Keeling J, Darby G, Jones Y, Stuart D, Stammers D. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nature Struct. Biol. 1995;2:293–308. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]
- Rodgers DW, Gamblin SJ, Harris BA, Ray S, Culp JS, Hellmig B, Woolf DJ, Debouck C, Harrison SC. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA. 1995;92:1222–1226. doi: 10.1073/pnas.92.4.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MJ, Tanese N, Goff SP. Purification and characterization of murine retroviral reverse transcriptase expressed in Escherichia coli. J. Biol. Chem. 1985;260:9326–9335. [PubMed] [Google Scholar]
- Rothwell PJ, Berger S, Kensch O, Felekyan S, Antonik M, Wohrl BM, Restle T, Goody RS, Seidel CAM. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl Acad. Sci. USA. 2003;100:1655–1660. doi: 10.1073/pnas.0434003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Clark AD, Jr, Das K, Tuske S, Birktoft JJ, Ilankumaran P, Ramesha AR, Sayer J, Jerina DM, Boyer PL, Hughes SH, Arnold E. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 2002;21:6614–6624. doi: 10.1093/emboj/cdf637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Das K, Tantillo C, Arthur D, Clark J, Jianping Ding, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz SJ, Zhang M, Champoux JJ. Sequence, distance, and accessibility are determinants of 5′ end-directed cleavages by retroviral RNases H. J. Biol. Chem. 2006;281:1943–1955. doi: 10.1074/jbc.M510504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindyalov I, Bourne P. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Temiz N, Bahar I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr HIV Res. 2004;2:323–332. doi: 10.2174/1570162043351093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanese N, Goff SP. Domain structure of Moloney murine leukemia virus reverse transcriptase: Mutational analysis and separate expression of the DNA polymerase and RNase H activities. Proc. Natl. Acad. Sci. USA. 1988;85:1777–1781. doi: 10.1073/pnas.85.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanese N, Telesnitsky A, Goff SP. Abortive reverse transcription by mutants of Moloney murine leukemia virus deficient in the reverse transcriptase-associated RNase H function. J Virol. 1991;65:4387–4397. doi: 10.1128/jvi.65.8.4387-4397.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, Blain SW, Goff SP. Defects in Moloney murine leukemia virus replication caused by a reverse transcriptase mutation modeled on the structure of Escherichia coli RNase H. J. Virol. 1992;66:615–622. doi: 10.1128/jvi.66.2.615-622.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, Goff SP. RNase H domain mutations affect the interaction between Moloney murine leukemia virus reverse transcriptase and its primer-template. Proc. Natl. Acad. Sci USA. 1993;90:1276–1280. doi: 10.1073/pnas.90.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiz NA, Bahar I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins: Structure, Function, and Genetics. 2002;49:61–70. doi: 10.1002/prot.10183. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Gorelick R. Nucleocapsid protein function in early infection processes. Virus Res. 2008 doi: 10.1016/j.virusres.2007.12.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale M, Schulze T, Larder BA, Moelling K. Mutations within RNase H domain of HIV-1 reverse transcriptase abolish virus infectivity. J. Gen. Virol. 1991;72:59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- Tsunaka Y, Takano K, Matsumura H, Yamagata Y, Kanaya S. Identification of single Mn(2+) binding sites required for activation of the mutant proteins of E.coli RNase HI at Glu48 and/or Asp134 by X-ray crystallography. J Mol Biol. 2005;345:1171–1183. doi: 10.1016/j.jmb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Tuske S, Sarafianos SG, Clark AD, Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat Struct Mol Biol. 2004;5:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- Wapling J, Moore K, Sonza S, Mak J, Tachedjian G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J. Virol. 2005;79:10247–10257. doi: 10.1128/JVI.79.16.10247-10257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]