

Trigonal crystals of MJ0729 showing different degrees of merohedral twinning that may vary from perfect hemihedral twinning to perfect tetartohedral twinning were obtained upon slight variation of the pH.
Keywords: MJ0729, merohedral twinning, CBS domain
Abstract
CBS domains are small protein motifs, usually associated in tandem, that are implicated in binding to adenosyl groups. Several genetic diseases in humans have been associated with mutations in CBS sequences, which has made them very promising targets for rational drug design. Trigonal crystals of the CBS-domain protein MJ0729 from Methanococcus jannaschii were grown by the vapour-diffusion method at acidic pH. Preliminary analysis of nine X-ray diffraction data sets using Yeates statistics and Britton plots showed that slight variation in the pH as well as in the buffer used in the crystallization experiments led to crystals with different degrees of merohedral twinning that may vary from perfect hemihedral twinning to perfect tetartohedral twinning.
1. Introduction
CBS domains are small motifs (Zhang et al., 1999 ▶) that are widely present in proteins of organisms from all kingdoms of life (InterPro IPR000644). Two or four motifs are usually associated, generating a simple compact structure that was first described in inosine monophosphate dehydrogenase IMPDH (Zhang et al., 1999 ▶). However, the function of CBS domains within cell biology remains poorly understood. In humans, CBS domains have been detected in a variety of cytoplasmic and nuclear proteins and carry out a wide range of molecular functions (http://www.ensembl.org). Several pathologies associated with mutations within CBS domains have been described: homocystinuria, autosomic retinitis pigmentosa, myotonia congenita, idiopathic epilepsy, hypercalciuric nephrolytiasis, classic Bartter’s syndrome, osteopetrosis, familiar hypertrophic cardiomyopathy and Wolff–Parkinson–White syndrome. This makes these domains promising targets for the development of novel drugs (Ignoul & Eggermont, 2005 ▶). The recently published crystal structures of AMP-dependent protein kinase (Xiao et al., 2007 ▶; Day et al., 2007 ▶; Jin et al., 2007 ▶; Amodeo et al., 2007 ▶; Townley & Shapiro, 2007 ▶) and of chloride channels (Meyer et al., 2007 ▶) have provided crucial information for understanding CBS–nucleotide binding and possible modes of function. As some hyperthermophile microorganisms from archaea, including Methanococcus jannaschii (Bult et al., 1996 ▶), possess a high number of proteins with CBS domains in their genome, they are excellent models for the study and characterization of the binding sites for different types of adenosyl groups in CBS domains, as well as for the study of the specificity of these proteins for their ligands.
We have crystallized the recombinant protein MJ0729 from M. jannaschii. The ORF of gene mj0729 (UniProtKB/Swiss-Prot Q58139) codes for a polypeptide chain of 124 amino acids with a molecular weight of 14.303 kDa. Its sequence consists of a CBS-domain pair (CBS1, residues 13–60; CBS2, residues 73–122; http://smart.embl-heidelberg.de/). Preliminary analysis of nine X-ray data sets collected from trigonal crystals indicated that they were twinned and may be affected by perfect hemihedral to perfect tetartohedral twinning. Twinning is a crystal-growth defect that frequently arises when two or more domains within a crystal have different orientations that are related to each other by a specific symmetry operation (Parsons, 2003 ▶). Consequently, it often has severe consequences for structure determination and refinement. If the lattices of the individual crystals overlap perfectly in the three dimensions, the diffraction pattern is a superposition of the diffraction pattern of each individual twin domain and the larger aggregate that is merohedrally twinned. Most merohedrally twinned protein crystals reported in the literature consist of two domains and these are called hemihedral twins. The twinning is perfect when the value of the twin ratio is close to 0.5. In this case, proper crystallographic intensities cannot be extracted from diffraction data. The structure can still be solved (Breyer et al., 1999 ▶; Dauter, 2003 ▶; Terwisscha van Scheltinga et al., 2003 ▶); however, the data set contain two times fewer independent reflections. A more complicated form of merohedral twinning is tetartohedral twinning, which has only been reported in three cases for protein crystals (Rosendal et al., 2004 ▶; Barends et al., 2005 ▶; Gayathri et al., 2007 ▶). In fact, tetartohedral twinning is considered to be an obscure rarity in protein crystallography (Barends et al., 2005 ▶). In this case, four twinning fractions and three twinning operators are defined. If tetartohedral twinning is strong extra symmetry in the diffraction data is observed, as in the case described by Rosendal et al. (2004 ▶), in which tetartohedral twinning added 222 symmetry to the P3 space-group symmetry, resulting in apparent 622 point-group symmetry of the crystal (Rosendal et al., 2004 ▶). Some authors have pointed out that only particles with 222 point-group symmetry are likely to yield tetartohedrally twinned crystals (Rosendal et al., 2004 ▶). However, the crystal structure of rat apo haem oxygenase-1 (PDB code 1irm; Sugishima et al., 2002 ▶), in which a P32 network is filled with additional rows of molecules at the positions of the threefold axis, is apparently a counterexample to this statement.
Our preliminary analysis of nine X-ray diffraction data sets using Yeates statistics and Britton plots showed that slight variations in the pH as well as in the buffer used in the crystallization experiments led to crystals with different degrees of merohedral twinning that may vary from perfect hemihedral twinning to perfect tetartohedral twinning.
2. Cloning and expression
The gene for MJ0729 was PCR-amplified with Pfu polymerase (Invitrogen) using the end primers 5′-CACCATGATGATAATGAAAG and 3′-CTATCTATTTTTATAATA (Sigma). The M. jannaschii genomic DNA used as the template was obtained from Sung-Hou Kim, University of Berkeley, California, USA. Gene mj0729 was cloned into the Escherichia coli expression vector pET-101D (Invitrogen) and was confirmed by DNA sequencing. The ligated plasmid was transformed into chemically competent E. coli BL21 (DE3) supplemented with plasmid pSJS1244 containing some of the rare E. coli tRNA genes (Kim et al., 1998 ▶). E. coli cells transformed with mj0729 were induced using 0.1–1 mM isopropyl β-d-1-thiogalactopyranoside at an OD600nm of 0.6 and were harvested by centrifugation at 5624g for 30 min. Test expression of the protein showed that E. coli overexpresses protein that is soluble and heat-stable. Strong leaky expression in the absence of IPTG led us not to use IPTG induction in subsequent experiments. A freshly transformed colony was transferred into 1 l LB broth (with 100 µg ml−1 ampicillin and 3 µg ml−1 spectinomicin) and grown overnight at 310 K to saturation. This overnight culture was used to inoculate 4 l LB broth (with antibiotics) and grown overnight at 310 K.
3. Protein purification
The cells pellet was resuspended in 20 ml lysis buffer containing 100 mM Tris pH 8.0, 1 mM EDTA, 0.1 mM PMSF and 1 mM benzamidine before being disrupted by sonication. The lysate was centrifuged at 145 000g for 30 min at 277 K to remove cell debris. After precipitation with 0.1% PEP (polyethylenimine P), the supernatant was heated at 358 K for 1 h and then centrifuged at 3901g for 15 min in an SX4250 rotor (Beckman). The supernatant was loaded onto a HiTrapQ 5 ml column (GE Healthcare) equilibrated with buffer A (50 mM HEPES pH 7.0, 1 mM EDTA, 1 mM BME) and connected to an ÄKTA-Basic (GE Healthcare) FPLC system. The protein was eluted with a linear salt gradient from 0 to 1 M NaCl in the same buffer at a flow rate of 1 ml min−1 for 45 min. Fractions containing MJ0729 (around 400 mM NaCl) were pooled and diluted three times in buffer A. They were reloaded onto the same column, but this time the protein was eluted using a segmented gradient at the same flow rate from 0 to 400 mM NaCl. MJ0729 was concentrated by ultrafiltration with Vivaspin 5K filters (Sartorius) to a final concentration of approximately 1 mg ml−1. In a final polishing step, concentrated MJ0729 was applied onto a high-resolution gel-filtration column (Superdex-75 HiLoad 16/60 prep grade, GE Healthcare) equilibrated with buffer containing 50 mM HEPES pH 7.0, 200 mM NaCl, 1 mM EDTA and 1 mM BME at a flow rate of 0.5 ml min−1. The Superdex-75 column was calibrated with lysozyme (14.6 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa) and ovoalbumin (44.3 kDa) as standards. Three peaks corresponding to tetrameric, dimeric and monomeric forms of MJ0729 were detected. Fractions corresponding to these three peaks were loaded onto an SDS–PAGE gel and bands corresponding to the molecular weight of MJ0729 were isolated and confirmed by mass spectrometry. Fractions containing each of the oligomers were dialyzed extensively against buffer containing 50 mM HEPES pH 7.0, 1 mM BME and 1 mM EDTA and were concentrated independently using a Vivaspin 5K filter (Sartorius) for crystallization trials. The concentration of the protein was estimated using the Bradford assay.
4. Mass-spectrometric analysis
SDS–PAGE gel bands were subjected to in-gel tryptic digestion according to Shevchenko et al. (1996 ▶) with minor modifications. The gel piece was swollen in a digestion buffer containing 50 mM NH4HCO3 and 12.5 ng µl−1 trypsin (Roche Diagnostics) in an ice bath. After 30 min, the supernatant was removed and discarded, 20 µl 50 mM NH4HCO3 was added to the gel piece and digestion was allowed to proceed at 310 K overnight. Prior to mass-spectrometric analysis, the sample was acidified by adding 5 µl 0.5% TFA. 0.5 µl digested sample was directly spotted onto the MALDI target and then mixed with 0.5 µl α-cyano-4-hydroxycinnamic acid (CHCA) matrix solution [20 µg µl−1 in acetonitrile, 0.1% TFA, 70:30(v:v)]. Peptide mass fingerprinting was performed on a Bruker Autoflex III mass spectrometer (Bruker-Daltonics, Bremen, Germany). Positively charged ions were analyzed in reflector mode using delayed extraction. The spectrum was obtained by randomly scanning the sample surface. About 600–800 spectra were averaged to improve the signal-to-noise ratio. Spectra were externally calibrated, resulting in a mass accuracy of <50 p.p.m. when external calibration was performed and typically <20 p.p.m. in the case of internal calibration. Protein identification was performed by searching in a nonredundant protein database (NCBI) using the Mascot search engine (http://matrixscience.com). The following parameters were used for database searches: one missed cleavage with allowed modifications carbamidomethylation of cysteine (complete) and oxidation of methionine (partial). Ultimately, MJ0729 expressed in E. coli was wild type and did not contain any mutations in the amino-acid sequence according to the M. jannaschii genome-sequence database.
5. Crystallization
Initial crystallization screening using the hanging-drop vapour-diffusion technique was carried out with a variety of commercial screens from Hampton Research (Crystal Screens 1 and 2, PEG/Ion Screen and SaltRX), mixing equal volumes (0.5 µl) of protein solution (in 100 mM HEPES pH 7.0, 1 mM BME, 1 mM EDTA) and reservoir solution. Protein concentrations ranging from 10 to 80 mg ml−1 (in 100 mM HEPES pH 7.0, 1 mM BME, 1 mM EDTA buffer) were tested. Needle-shaped crystals were obtained from condition No. 47 of Crystal Screen 1 (1 M ammoniun sulfate, 0.1 M sodium acetate pH 4.6) in less than 1 d at 295 K. From the initial experiments, it became clear that the shape and quality of the crystals varied significantly upon small variations in the pH. Consequently, further optimization of the crystallization conditions required accurate control of the pH by carefully adjusting the concentrations used in both the mother liquor and reservoir solution. Mixed solutions of mother liquor (25–100 mM HEPES pH 7.0, 1 mM BME, 1 mM EDTA) with reservoir solution 25–100 mM citric acid (or sodium acetate) pH 4.8–4.9, 1.4–1.6 M ammonium sulfate and a protein concentration of 55–65 mg ml−1 yielded the best diffraction-quality crystals, which typically grew in 1–3 d to maximum dimensions of 400 × 400 × 1500 µm. Three different crystal shapes were obtained: rocket-shaped rods with a hexagonal base (at pH 4.2–4.3; Fig. 1 ▶ a), sharp rods with a hexagonal base (at pH 4.6–4.8; Fig. 1 ▶ b) and hexagonal prisms (at pH 4.8–5.0; Fig. 1 ▶ c). Dynamic light-scattering (DLS) analysis showed that MJ0729 was a tetramer in solution in the pH range used for crystallization (pH 4.2–5.0). Good-quality crystals, especially those with the rocket-like shape, were often very difficult to reproduce. Crystallization success was found to be highly dependent on the purification batch used. Very often, a shower of useless broom-shaped crystals formed of clusters of fine rods were obtained and pH adjustment was required to improve their quality. Prior to data collection, the crystals were transferred to crystallization buffer containing 30% glycerol as cryoprotectant for a few seconds before being flash-cooled by direct immersion into liquid nitrogen at 93 K.
Figure 1.

Trigonal crystals of MJ0729 grown by vapour diffusion at 293 K and (a) pH 4.2–4.3 (rocket-shaped rods with a hexagonal base), (b) pH 4.6–4.8 (sharp rods with a hexagonal base) and (c) pH 4.8–5.0 (hexagonal prisms) (approximate dimensions 400 × 400 × 1500 µm). κ = 180° sections of the self-rotation function of the native MJ0729 data sets RAN1, BM2 and ID1 are shown in (d), (e) and (f), respectively. The apparent 321 point-group symmetry observed for data set RAN1 is consistent with both a nontwinned P3121/P3221 crystal or with an almost perfect hemihedrally twinned P31/P32 crystal. On the other hand, the apparent 622 point-group symmetry observed for data sets BM2 and ID1 suggests different degrees of hemihedral or tetartohedral twinning, depending on the actual space group (P3121/P3221 or P31/P32, respectively). This figure was prepared using MOLREP (Vagin & Teplyakov, 1998 ▶).
6. Data collection and processing
Eight X-ray diffraction data sets were collected at synchrotron beamlines BM16 and ID14-1 at the European Synchrotron Research Facility (ESRF), Grenoble, France. An additional data set (RAN1) was collected at 100 K on a 135 mm CCD detector (Bruker) using an X8-Proteum System equipped with a rotating-anode X-ray source operated at 45 kV and 60 mA with Cu Kα radiation focused using a Helios mirror system. Crystals of native MJ0729 diffracted X-rays to resolution limits ranging from 4 to 2.3 Å. Data sets were processed using MOSFLM (Leslie, 2006 ▶) and/or the HKL-2000 package (Otwinowski & Minor, 1997 ▶; Table 1 ▶). In all cases the diffraction spots were clearly defined (see Fig. 2 ▶).
Table 1. Data-processing statistics for native MJ0729.
Twinning fractions were calculated for the three possible operators (TO1, TO2 and TO3) in space group P31 using the Yeates method (Yeates, 1997 ▶). BM and ID data sets were collected at synchrotron beamlines BM16 and ID23, respectively, at the European Synchrotron Research Facility (ESRF), Grenoble, France. Data set RAN1 was collected on a 135 mm CCD detector (Bruker) using FR591 rotating-anode Cu Kα radiation.
| Data set | RAN1 | BM2 | BM3 | BM8 | BM9 | BM10 | ID1 | ID2 | ID3 |
|---|---|---|---|---|---|---|---|---|---|
| Beamline | FR591 | BM16 | BM16 | BM16 | BM16 | BM16 | ID23-1 | ID23-1 | ID23-1 |
| Wavelength (Å) | 1.54 | 0.9797 | 0.9797 | 0.9797 | 0.9797 | 0.9797 | 0.9340 | 0.9340 | 0.9340 |
| No. of observations | 17627 | 50588 | 39361 | 37991 | 63606 | 62760 | 526547 | 345870 | 369863 |
| No. of unique reflections | 6055 | 17597 | 10551 | 11233 | 16355 | 16069 | 35292 | 28133 | 25108 |
| Resolution range (Å) | 38–4.2 | 3.1 | 3.5 | 58–3.4 | 58–3.2 | 64–3.0 | 36–2.3 | 50–2.5 | 50–2.6 |
| Space group | P31/P32 or P3121/P3221 | ||||||||
| Unit-cell parameters | |||||||||
| a (Å) | 75.431 | 73.493 | 75.043 | 74.333 | 74.470 | 74.224 | 73.186 | 73.006 | 72.829 |
| b (Å) | 75.431 | 73.493 | 75.043 | 74.333 | 74.470 | 74.224 | 73.186 | 73.006 | 72.829 |
| c (Å) | 132.211 | 131.974 | 131.512 | 131.920 | 131.919 | 131.880 | 132.342 | 132.431 | 132.762 |
| Rmerge† | 0.200 | 0.083 | 0.098 | 0.150 | 0.142 | 0.136 | 0.063 | 0.043 | 0.064 |
| Completeness (%) | 98.8 | 99.8 | 99.7 | 100.0 | 99.8 | 98.7 | 98.7 | 99.5 | 99.8 |
| Mean I/σ(I) | 6.8 | 14.4 | 9.55 | 9.3 | 11.6 | 12.3 | 30.81 | 42.62 | 32.89 |
| Mosaicity (°) | 0.50 | 0.80 | 2.00 | 0.50 | 0.49 | 0.60 | 0.76 | 0.40 | 0.69 |
| Wilson B factor (Å2) | 28.85 | 72.31 | 53.54 | 46.58 | 64.95 | 75.17 | 54.432 | 54.46 | 68.58 |
| Redundancy | 2.9 | 2.9 | 3.7 | 3.4 | 3.9 | 3.9 | 3.1 | 3.2 | 6.5 |
| Twinning fraction (in P3) | |||||||||
| TO1 = (h, −h − k, −l) | 0.388 | 0.446 | 0.455 | 0.410 | 0.434 | 0.430 | 0.489 | 0.484 | 0.487 |
| TO2 = (h + k, −k, −l) | 0.008 | 0.144 | 0.385 | 0.057 | 0.241 | 0.220 | 0.431 | 0.431 | 0.233 |
| TO3 = (−h, −k, l) | 0.029 | 0.155 | 0.388 | 0.070 | 0.249 | 0.225 | 0.432 | 0.432 | 0.238 |
R
merge = 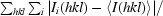
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Figure 2.

Representative X-ray diffraction image from MJ0729 (data set ID1). The crystal was exposed for 5 s over a 1° oscillation range. The edge of the detector corresponds to a resolution of 1.7 Å.
7. Detection and analysis of twinning in the crystal
Twinning was first suggested by the detection of differences in the heights of the twofold peaks perpendicular to the threefold axis in the self-rotation function (SRF) of several data sets. According to their apparent point-group symmetry deduced from the SRF (Fig. 1 ▶), the space group for the BM and ID data sets was initially determined as P622 or any of its variants with one molecule per asymmetric unit and a V M of 3.58 Å3 Da−1 (Matthews, 1968 ▶). In the same way, the space group for data set RAN1 was determined as P321 or any of its variants with two molecules per asymmetric unit. Detailed analysis of the systematic absences (l = 3n) along the reciprocal-space vector 00l indicated space group P6422 or its enantiomorph P6222 for the BM and ID data sets, and space group P3121 (or its enantiomorph P3221) for data set RAN1. The solvent content (approximately 66%) was rather high in all cases. However, after processing the data sets in all possible trigonal and hexagonal point groups (Table 2 ▶) to aid in the detection of pseudo-symmetry, it became evident that decreasing the crystal symmetry to trigonal P3 or P321 improved the data-proccessing statistics in all cases apart from data set ID2, for which no variation was observed (Table 2 ▶). Accordingly, the number of molecules in the asymmetric unit was estimated to be two (in P3 groups) or four (in P321 groups).
Table 2. R merge values for MJ0729 data sets proccessed in all possible point groups compatible with trigonal and hexagonal lattices.
R
merge =
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
| P3 | P321 | P | P6 | P622 | |
|---|---|---|---|---|---|
| RAN1 | 0.200 | 0.324 | 0.447 | 0.519 | 0.538 |
| BM2 | 0.083 | 0.080 | 0.236 | 0.241 | 0.255 |
| BM3 | 0.098 | 0.105 | 0.136 | 0.137 | 0.140 |
| BM8 | 0.150 | 0.165 | 0.414 | 0.379 | 0.439 |
| BM9 | 0.142 | 0.149 | 0.274 | 0.272 | 0.276 |
| BM10 | 0.136 | 0.146 | 0.285 | 0.284 | 0.289 |
| ID1 | 0.063 | 0.065 | 0.090 | 0.090 | 0.091 |
| ID2 | 0.043 | 0.045 | 0.045 | 0.045 | 0.046 |
| ID3 | 0.064 | 0.065 | 0.268 | 0.268 | 0.270 |
The statistics shown in Table 2 ▶ suggest that data set RAN1 may correspond to a hemihedrally twinned crystal with symmetry P31 or P32. However, at this point it is difficult to unambiguously determine the actual space group, since RAN1 has lower resolution and poorer statistics than the other data sets. The BM and ID data sets may correspond to either P3 or P321 groups.
Since trigonal space groups are prone to twinning, native data sets were also tested for twinning. The intensity statistics, including the cumulative intensity plot and the moments of normalized intensities (Stanley, 1972 ▶; Dauter, 2003 ▶), were obtained using the Twinning Server (http://www.doe-mbi.ucla.edu/Services/Twinning; Yeates, 1997 ▶), the program TRUNCATE from CCP4 (Collaborative Computational Project, Number 4, 1994 ▶) and the program CNS (Brünger et al., 1998 ▶). The local intensity statistics (Padilla & Yeates, 2003 ▶) were analyzed using the program DATAMAN (Kleywegt & Jones, 1996 ▶). The twinning fraction was initially estimated from the cumulative distribution of H (Yeates, 1997 ▶) and Britton plots (Fisher & Sweet, 1980 ▶; Britton, 1972 ▶) with the CCP4 program DETWIN (Collaborative Computational Project, Number 4, 1994 ▶). Statistical analysis of the intensities clearly indicated that the crystals used for data collection were merohedrally twinned (Table 1 ▶). Native Patterson maps did not contain any significant peaks that might have indicated the presence of pseudo-translation. Twinning could also be detected from the corresponding self-rotation functions (SRFs), similar to the determination of noncrystallographic symmetry. The relative height of the extra peaks compared with the peaks related to crystallographic symmetry is indicative of the importance of twinning. The SRFs (Fig. 1 ▶) were calculated with the program MOLREP (Collaborative Computational Project, Number 4, 1994 ▶). In addition to the crystallographic threefold axis, the SRFs show an additional twofold axis parallel to the c axis and also along a*, b* and along the a and b axes (Fig. 1 ▶). The relative ratio of the crystallographic threefold peak to the twofold peaks induced by the twinning series along the a, b and a*, b* axes varies from 0 to 98% for the nine data sets, indicating a broad range of twinning (Table 1 ▶). This ratio always remains close to 100% in all data sets for the twofold peak parallel to the c axis, suggesting that its presence arises from twinning (twofold along c) or from twofold NCS internal symmetry in the molecule. According to this data, the type of twinning observed in MJ0729 crystals seemed to range from pure hemihedral in data set RAN1 to perfect tetartohedral in ID1, ID2 and ID3. The BM data sets displayed an intermediate twinning ratio. As pointed out by other authors (Lee et al., 2003 ▶), the partial twinning test is not in itself proof of twinning. It cannot distinguish between NCS and twinning operations if the NCS axis happens to be close to a twinning operator (Yeates & Fam, 1999 ▶). When twinning is promoted by NCS, the effects of twinning on the intensity statistics are reduced and the averaging effect of the twinning is less pronounced. This happens as the reflections averaged by the twinning are already strongly correlated owing to the NCS (Stanley, 1972 ▶; Barends et al., 2005 ▶).
From the data shown here, we cannot exclude the possibility that twinning operator TO1 is a crystallographic operator, since the contrast in R merge is marginal for all data sets (except for the low-resolution data set RAN1; Tables 1 ▶ and 2 ▶) and no distinction between NCS and twinning can be made based on the SRF. Further studies in the context of structural determination are in progress to unambiguously determine the actual space-group symmetry using a comparative analysis of statistics based on observed and calculated intensities. This will also determine the actual number of crystal domains (two or four) that contribute to the corresponding diffraction patterns.
Acknowledgments
We thank Professor Sung-Hou Kim of University of California at Berkeley for providing us with the genomic DNA from M. jannaschii and the staff of ESRF beamlines BM16 and ID14-1 for support during synchrotron data collection. We are grateful to Professor Martin Martínez-Ripoll for helpful discussions. We also thank Beatriz González Callejas from CIC bioGUNE for maintenance of the in-house X-ray equipment. This research was supported by program grants from NIH, the Basque Government (Etortek) and the Spanish Ministry of Education (MEC) and predoctoral and postdoctoral fellowships from CIC bioGUNE.
References
- Amodeo, G. A., Rudolph, M. J. & Tong, L. (2007). Nature (London), 449, 492–495. [DOI] [PubMed]
- Barends, T. R. M., de Jong, R. M., van Straaten, K. E., Thunnissen, A.-M. W. H. & Dijkstra, B. W. (2005). Acta Cryst. D61, 613–621. [DOI] [PubMed]
- Breyer, W. A., Kingston, R. L., Anderson, B. F. & Baker, E. N. (1999). Acta Cryst. D55, 129–138. [DOI] [PubMed]
- Britton, D. (1972). Acta Cryst. A28, 296–297.
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bult, C. J. et al. (1996). Science, 273, 1058–1073. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dauter, Z. (2003). Acta Cryst. D59, 2004–2016. [DOI] [PubMed]
- Day, P., Sharff, A., Parra, L., Cleasby, A., Williams, M., Hörer, S., Nar, H., Redemann, N., Tickle, I. & Yon, J. (2007). Acta Cryst. D63, 587–596. [DOI] [PubMed]
- Fisher, R. G. & Sweet, R. M. (1980). Acta Cryst. A36, 755–760.
- Gayathri, P., Banerjee, M., Vijayalakshmi, A., Azeez, S., Balaram, H., Balaram, P. & Murthy, M. R. N. (2007). Acta Cryst. D63, 206–220. [DOI] [PubMed]
- Ignoul, S. & Eggermont, J. (2005). Am. J. Physiol. Cell Physiol.289, 1369–1378. [DOI] [PubMed]
- Jin, X., Townley, R. & Shapiro, L. (2007). Structure, 15, 1285–1295. [DOI] [PubMed]
- Kim, R., Sandler, S. J., Goldman, S., Yokota, H., Clark, A. J. & Kim, S.-H. (1998). Biotechnol. Lett.20, 207–210.
- Kleywegt, G. J. & Jones, T. A. (1996). Acta Cryst. D52, 826–828. [DOI] [PubMed]
- Lee, S., Sawaya, M. R. & Eisenberg, D. (2003). Acta Cryst. D59, 2191–2199. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Meyer, S., Savaresi, S., Forster, I. C. & Dutzler, R. (2007). Nature Struct. Mol. Biol.14, 60–67. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Padilla, J. E. & Yeates, T. O. (2003). Acta Cryst. D59, 1124–1130. [DOI] [PubMed]
- Parsons, S. (2003). Acta Cryst. D59, 1995–2003. [DOI] [PubMed]
- Rosendal, K. R., Sinning, I. & Wild, K. (2004). Acta Cryst. D60, 140–143. [DOI] [PubMed]
- Shevchenko, A., Wilm, M., Vorm, O. & Mann, M. (1996). Anal. Chem.68, 850–858. [DOI] [PubMed]
- Stanley, E. (1972). J. Appl. Cryst.5, 191–194.
- Sugishima, M., Sakamoto, H., Kakuta, Y., Omata, Y., Hayashi, S., Noguchi, M. & Fukuyama, K. (2002). Biochemistry, 41, 7293–7300. [DOI] [PubMed]
- Terwisscha van Scheltinga, A. C., Valegård, K., Hajdu, J. & Andersson, I. (2003). Acta Cryst. D59, 2017–2022. [DOI] [PubMed]
- Townley, R. & Shapiro, L. (2007). Science, 315, 1726–1729. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1998). Acta Cryst. D54, 400–402. [DOI] [PubMed]
- Xiao, B., Heath, R., Saiu, P., Leiper, F. C, Leone, P., Jing, C., Walker, P. A., Haire, L., Eccleston, J. F., Davis, C. T., Martin, S. R., Carling, D. & Gamblin, S. J. (2007). Nature (London), 449, 496–500. [DOI] [PubMed]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–358. [PubMed]
- Yeates, T. O. & Fam, B. C. (1999). Structure, 7, 25–29. [DOI] [PubMed]
- Zhang, R., Evans, G., Rotella, F. J., Westbrook, E. M., Beno, D., Huberman, E., Joachimiak, A. & Collart, F. R. (1999). Biochemistry, 38, 4691–4700. [DOI] [PubMed]