


Abstract
Given an mRNA sequence as input, the OligoWalk web server generates a list of small interfering RNA (siRNA) candidate sequences, ranked by the probability of being efficient siRNA (silencing efficacy greater than 70%). To accomplish this, the server predicts the free energy changes of the hybridization of an siRNA to a target mRNA, considering both siRNA and mRNA self-structure. The free energy changes of the structures are rigorously calculated using a partition function calculation. By changing advanced options, the free energy changes can also be calculated using less rigorous lowest free energy structure or suboptimal structure prediction methods for the purpose of comparison. Considering the predicted free energy changes and local siRNA sequence features, the server selects efficient siRNA with high accuracy using a support vector machine. On average, the fraction of efficient siRNAs selected by the server that will be efficient at silencing is 78.6%. The OligoWalk web server is freely accessible through internet at http://rna.urmc.rochester.edu/servers/oligowalk.
INTRODUCTION
It is well known that genes can be silenced by antisense RNA oligonucleotides called small interfering RNA (siRNA) (1,2). In order to design an efficient siRNA sequence, empirical rules based on the features of the siRNA sequence have been discovered, including, for example, low G/C content, lack of self-structure, preference of A at position 3, absence of G or C at position 19 and asymmetry in the stability of the terminal base pairs (3–10). The self-structure of the target and oligonucleotide is also an important consideration for the effective binding (11–15). It is desirable to select an oligonucleotide having high accessibility to the target-binding site and low duplex stability. Here, the OligoWalk server, which predicts efficient siRNA sequences using an accessibility calculation with a convenient web interface, is described. Overall, the positive predictive value of the server is 0.786, meaning that 78.6% of the siRNAs selected by the server will be efficient at silencing (16). The positive predictive value was determined by testing against a database of siRNA experiments conducted under diverse experimental conditions (17).
In the calculation of the OligoWalk server, unimolecular and bimolecular self-structures for the siRNA are considered along with unimolecular self-structure in the target at the oligonucleotide binding region (16). These structures are in equilibrium with each other and with the hybridized state. OligoWalk predicts the free energy changes (ΔG◦) involved in these equilibrium states (18). The predicted thermodynamics (ΔG◦), plus the oligonucleotide sequence features (19), are then utilized to predict siRNA efficacy for candidate siRNA sequences (16), which are generally 19 nucleotide duplexes with 3′ dinucleotide dangling ends (7). A support vector machine (SVM) program (20) is embedded in the server to take the thermodynamic and sequence features as input. The SVM classification model used in the server has been proven to be able to predict efficient siRNA (greater than 70% inhibition of the target mRNA expression) with high accuracy (16). The SVM was trained on a siRNA database that contains 2431 experimental results conducted in human cells at 37°C (10).
The input is the sequence of the target RNA. Advanced options are available for expert users to customize the calculation. The output of the OligoWalk server is a table of siRNA candidates, showing the siRNA sequences and the probabilities of being efficient (having silencing efficacy larger than 70%). Each of the free energy change terms for each candidate is also listed in a separate table.
OLIGOWALK SERVER INPUT
The OligoWalk server uses the CGI (Common Gateway Interface) module of Perl for taking user input and submitting calculations from the homepage. The input of OligoWalk server is the RNA sequence of the target gene. Only A, U, T, G and C are the acceptable types of nucleotides in the sequence (the server will replace the nucleotide T with U for calculations), and the maximum sequence length is 10 000 nucleotides. An email address is required because the server sends an email to the user when the calculation is completed. Online help is available at the ‘Help’ hyperlink. When the user clicks, ‘Submit Query’, the server generates a list of efficient siRNA candidates for the target gene. Jobs are submitted by the server to a cluster of seven nodes with 3.2 or 3.4 GHz Pentium 4 processors running Fedora Linux (http://fedoraproject.org/), managed by Sun Grid Engine (http://gridengine.sunsource.net/). The default siRNA candidate is an RNA oligonucleotide having 19 nucleotides.
OLIGOWALK SERVER OUTPUT
When the calculation is complete, an html (hypertext markup language) page is generated with links to tables containing predicted siRNA efficacy data and thermodynamic binding data. In the siRNA efficacy table (Figure 1), the sequences of siRNA candidates are ranked in the output list by their probabilities of being efficient siRNA. The probabilities are predicted by a SVM embedded in the web server for selecting efficient siRNA. The classification model (16) used in the SVM was trained with a publically available database (10), using thermodynamic and sequence features of siRNA candidates. The position number of each siRNA candidate is also listed in the table as the index of the 5′ most base in the target-binding region.
Figure 1.

A table of siRNA candidates generated by OligoWalk server. The sequences of siRNA are ranked from top to end by their probabilities of being efficient (antisense efficacy larger than 70%).
In addition, the predicted equilibrium thermodynamics table is generated as a reference for advanced users. In the table, the position number and sequence of each siRNA candidate appear with thermodynamic terms. ‘Overall’ (in kcal/mol) is the overall free energy change of oligonucleotide-target binding, 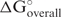 when all contributions are considered, including breaking target and oligonucleotide self-structures (18). A more negative value indicates tighter binding. It is affected by the oligonucleotide concentration. ‘Duplex’ (in kcal/mol) is the free energy change of hybridized duplex between oligonucleotide and target (antisense–sense duplex),
when all contributions are considered, including breaking target and oligonucleotide self-structures (18). A more negative value indicates tighter binding. It is affected by the oligonucleotide concentration. ‘Duplex’ (in kcal/mol) is the free energy change of hybridized duplex between oligonucleotide and target (antisense–sense duplex), 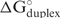 . The value is independent of oligonucleotide concentration because it is a standard free energy change. ‘Tm-Dup’ (in °C) is the melting temperature in degrees for the duplex formation of oligonucleotide and target. ‘Break-targ’. (in kcal/mol) is the free energy cost to open the intramolecular target base pairs for oligonucleotide binding,
. The value is independent of oligonucleotide concentration because it is a standard free energy change. ‘Tm-Dup’ (in °C) is the melting temperature in degrees for the duplex formation of oligonucleotide and target. ‘Break-targ’. (in kcal/mol) is the free energy cost to open the intramolecular target base pairs for oligonucleotide binding, 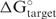 . A more negative number indicates higher free energy cost, which is unfavorable for oligonucleotide-target binding. ‘Intraoligo’ (in kcal/mol) is the free energy change of intramolecular oligonucleotide structure,
. A more negative number indicates higher free energy cost, which is unfavorable for oligonucleotide-target binding. ‘Intraoligo’ (in kcal/mol) is the free energy change of intramolecular oligonucleotide structure, 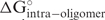 . It usually has a negative value or, if there is no favor-able intramolecular structure, it is zero. ‘Interoligo’ (in kcal/mol) is the free energy change of intermolecular oligonucleotide structure,
. It usually has a negative value or, if there is no favor-able intramolecular structure, it is zero. ‘Interoligo’ (in kcal/mol) is the free energy change of intermolecular oligonucleotide structure, 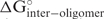 . A negative number indicates a stable antisense–antisense bimolecular structure, which decreases the oligonucleotide-target (antisense–sense) binding affinity. ‘End_diff’. (in kcal/mol) is the free energy difference between the 5′ and 3′ end of the antisense strand of siRNA, with windows of two base pairs. Functional siRNA prefer to have an unstable 5′ end (3), which means a positive End_diff. ‘Prefilter_score’ is the score calculated with a method based on the empirical rules by Reynolds et al. (7). All the scores are calculated in the same way as Reynolds et al. (7), except for the melting temperature of intramolecular oligonucleotide self-structure because the free energy (21) and enthalpy parameters (22) used by OligoWalk are more recent. When calculating the prefilter score, 57°C is used as the cutoff of the intramolecular oligonucleotide melting temperature, as suggested in another study (23).
. A negative number indicates a stable antisense–antisense bimolecular structure, which decreases the oligonucleotide-target (antisense–sense) binding affinity. ‘End_diff’. (in kcal/mol) is the free energy difference between the 5′ and 3′ end of the antisense strand of siRNA, with windows of two base pairs. Functional siRNA prefer to have an unstable 5′ end (3), which means a positive End_diff. ‘Prefilter_score’ is the score calculated with a method based on the empirical rules by Reynolds et al. (7). All the scores are calculated in the same way as Reynolds et al. (7), except for the melting temperature of intramolecular oligonucleotide self-structure because the free energy (21) and enthalpy parameters (22) used by OligoWalk are more recent. When calculating the prefilter score, 57°C is used as the cutoff of the intramolecular oligonucleotide melting temperature, as suggested in another study (23).
As an example, the prediction of the webserver is compared with experimental results in Figure 2. In the experiment (3), siRNA were tested for efficacy against the target mRNA, Human Cyclophilin (Genbank ID: M60857), at 37°C. The inhibition efficacy of each siRNA is defined as 100% minus the percentage of mRNA level after siRNA application as compared to matched control. The prediction result is the probability of being efficient (having inhibition efficacy larger than 70%), which is calculated with the server. In Figure 2, most of the siRNA with high inhibition efficacy are predicted to have high probability of being efficient.
Figure 2.

A comparison between the prediction and experimental result. In blue is the experimental result (inhibition efficacy) for an mRNA target, Human Cyclophilin (Genbank ID: M60857), at 37°C (3). The inhibition efficacy is defined as 100% minus the percent level of mRNA expression after siRNA application as compared to a control. In red is the probability (ranging from 0–1) of being efficient (having inhibition efficacy larger than 70%), as predicted by OligoWalk. Position is the starting position of each siRNA on the target sequence.
ADVANCED OPTIONS
Advanced options (Figure 3) are available for users who understand the underlying calculations and would like to test novel hypotheses. The option form is written in html, embedded with JavaScript controlling the options so that they are context-aware. The oligonucleotide length and concentration can be user-defined. The oligonucleotide concentration does not affect the result of siRNA sequence design, because the inputs to the SVM are standard free energy changes, ΔG◦. Concentration changes do, however, alter the overall free energy change (), which is provided to the user with the thermodynamic details table. Three options are then available for the ‘Binding mode’ calculation. These control the calculation of the target structure opening cost. The fastest calculation is to not consider target structure. In this mode, the sense–antisense duplex free energy changes are calculated without considering the self-structures of the target. This mode is not recommended for an accurate siRNA prediction. It is more rigorous to consider the accessibility of the target and oligonucleotide self-structures because binding affinity is lost to open existing base pairs in the target and oligonucleotide. The second mode is to break local structure. In this case, the structure of target is fixed and only the base pairs on the binding site will be broken without refolding the global structure of target. The final and most rigorous mode is to refold the target RNA. In this mode, the RNA target is folded before oligonucleotide binding and refolded afterwards for each possible oligonucleotide to consider complete equilibration. This is the default mode for the server.
Figure 3.

Advanced input options of OligoWalk web server. The default setting of siRNA design is shown in the figure.
If the target RNA secondary structure is considered, three different prediction methods are available to calculate the free energy change of target self-structure. The first method is optimal structure prediction, where only the optimal structure (lowest free energy structure) of the target is considered to calculate the free energy cost of opening the base pairs of binding region. The second method considers a set of suboptimal structures to determine the free energy cost. Each structure's free energy cost is weighted according to the free energy change of the structure to arrive at the ensemble cost. For this option, at most 1000 suboptimal structures (within 10% free energy difference from the optimal structure) are generated with a heuristic method (24). The number of suboptimal structures will be listed in the output table if the target is folded with suboptimal structure prediction method. There are two columns of structure numbers in the output table. The first one is the number of target structures being predicted before oligonucleotide binding. The second one is the number of constrained target structures. Constrained target structure is the refolded structure where the binding region is forced to be single-stranded, so that the oligonucleotide can bind to it. The final and default option is a partition function calculation (25). This is the most rigorous method because it considers every possible secondary structure in the folding ensemble, with Boltzmann weight.
The structure prediction only folds a certain total number (folding size) of nucleotides centered at the binding region. The user can define this number, but the largest folding size is 1000 nucleotides for the webserver in order to save compute time. Users can define longer folding sizes by downloading and installing the OligoWalk program to a local machine. A prefilter based on the scoring method by Reynolds et al. (7) can be used to rule out nonefficient siRNA candidates before folding the target sequence, i.e. the siRNA sequences having score less than six points will not be considered for the folding step. It is suggested to turn on the prefilter option to save considerable computation time (Table 1). Furthermore, the scan region can be redefined if the user is interested only in a specific region of the target.
Table 1.
Calculation size and time of OligoWalk server for different target sequences
| Target mRNA (Genbank ID) | Sequence length (nucleotide) | Timea (h:min:sec) | Memory (MB) |
|---|---|---|---|
| NM_020548 | 730b | 0:57:17 | 93 |
| M60857 | 851 | 3:55:53 | 110 |
| NM_002870 | 1211 | 6:09:25 | 112 |
| NM_002467 | 2189 | 6:36:42 | 113 |
| AJ272212 | 3460 | 6:53:05 | 117 |
The benchmarks were performed with the default options: The oligonucleotide was a 19 base RNA; the folding size of the target was 800 nucleotides centering on the binding site (full length if the whole target has less than 800 nt); the partition function calculation was conducted; the entire mRNA was scanned; and the prefilter was on. The time cost changes little for long sequence because the prefilter (7) is on and number of candidates being folded is limited to about the same number for each sequence.
aThe calculations were submitted and benchmarked on the OligoWalk web server (http://rna.urmc.rochester.edu/servers/oligowalk). The cluster has up to seven executable nodes, managed by Sun Grid Engine. Each node has a 3.2 or 3.4 GHz Pentium 4 processor running Fedora Linux.
bThe calculation for sequences less than 800 nucleotides is relatively fast because the dynamic programming arrays are reused for calculations of short sequences (16).
CONCLUSIONS
The OligoWalk web server predicts the hybridization thermodynamics of an oligonucleotide binding to a complementary target RNA using the most recent RNA folding parameters (21, 22). It predicts efficient siRNA with high accuracy using a transparent implementation of an SVM (16), which considers both sequence and thermodynamic features. The calculation time and memory size of OligoWalk are shown in Table 1 for a sample of mRNA sequences. The prefilter (7) that uses local sequence information to narrow down the list of siRNA candidates before calculating the equilibrium affinity is used by default. Its use is recommended because the calculation of the partition function is time consuming. For example, the server takes 3 h and 43 min for a complete scan of all possible siRNAs on an mRNA having 730 nucleotides using the partition function calculation. For the same sequence, the time cost is only 57 min when the prefilter is turned on. The algorithm time scales O(mN 3) and the memory use scales O(N2) (Table 1), m is the number of candidates and N is the value of folding size. The time and memory costs change little with sequence because the same folding size (e.g. 800 nucleotides) is used and the prefilter (7) is turned on, which limits the number of candidates to be folded in a way that is apparently independent of target length.
There is currently significant interest in using siRNA for both basic science and medical research. The fact that not all siRNA duplexes will function in silencing means that there is a significant cost in trial and error for siRNA design. The OligoWalk server for siRNA design can mitigate this cost.
ACKNOWLEDGEMENTS
The design of the server was supported by the National Institutes of Health with grant R01GM076485 to D.H.M. Funding to pay the Open Access publication charges for this article was provided by the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Scherer LJ, Rossi JJ. Approaches for the sequence-specific knockdown of mRNA. Nat. Biotechnol. 2003;21:1457–1465. doi: 10.1038/nbt915. [DOI] [PubMed] [Google Scholar]
- 3.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 4.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 5.Amarzguioui M, Prydz H. An algorithm for selection of functional siRNA sequences. Biochem. Biophys. Res. Commun. 2004;316:1050–1058. doi: 10.1016/j.bbrc.2004.02.157. [DOI] [PubMed] [Google Scholar]
- 6.Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev. 2003;13:83–105. doi: 10.1089/108729003321629638. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall W.S, Khvorova A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 8.Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A, Ueda R, Saigo K. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32:936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, Meloon B, Engel S, Rosenberg A, Cohen D, et al. Design of a genome-wide siRNA library using an artificial neural network. Nat. Biotechnol. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- 11.Vickers TA, Wyatt JR, Freier SM. Effects of RNA secondary structure on cellular antisense activity. Nucleic Acids Res. 2000;28:1340–1347. doi: 10.1093/nar/28.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohula EA, Salisbury AJ, Sohail M, Playford MP, Riedemann J, Southern EM, Macaulay VM. The efficacy of small interfering RNAs targeted to the type 1 insulin-like growth factor receptor (IGF1R) is influenced by secondary structure in the IGF1R transcript. J. Biol. Chem. 2003;278:15991–15997. doi: 10.1074/jbc.M300714200. [DOI] [PubMed] [Google Scholar]
- 13.Far RK, Sczakiel G. The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res. 2003;31:4417–4424. doi: 10.1093/nar/gkg649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schubert S, Grunweller A, Erdmann VA, Kurreck J. Local RNA target structure influences siRNA efficacy: systematic analysis of intentionally designed binding regions. J. Mol. Biol. 2005;348:883–893. doi: 10.1016/j.jmb.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Heale BS, Soifer HS, Bowers C, Rossi JJ. siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res. 2005;33:e30. doi: 10.1093/nar/gni026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu ZJ, Mathews DH. Efficient siRNA selection using hybridization thermodynamics. Nucleic Acids Res. 2008;36:640–647. doi: 10.1093/nar/gkm920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shabalina SA, Spiridonov AN, Ogurtsov AY. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics. 2006;7:65. doi: 10.1186/1471-2105-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 1999;5:1458–1469. doi: 10.1017/s1355838299991148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ladunga I. More complete gene silencing by fewer siRNAs: transparent optimized design and biophysical signature. Nucleic Acids Res. 2007;35:433–440. doi: 10.1093/nar/gkl1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang C, Lin C. 2001. LIBSVM: a library for support vetor machines. http://www.csie.ntu.edu.tw/∼cjlin/libsvm/
- 21.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu ZJ, Turner DH, Mathews DH. A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res. 2006;34:4912–4924. doi: 10.1093/nar/gkl472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saetrom P, Snove O., Jr. A comparison of siRNA efficacy predictors. Biochem. Biophys. Res. Commun. 2004;321:247–253. doi: 10.1016/j.bbrc.2004.06.116. [DOI] [PubMed] [Google Scholar]
- 24.Zuker M. On finding all suboptimal foldings of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 25.Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]