

Abstract
NMR investigations have been carried out of complexes between bovine chymotrypsin Aα and a series of four peptidyl trifluoromethyl ketones, listed here in order of increasing affinity for chymotrypsin: N-Acetyl-l-Phe-CF3, N-Acetyl-Gly-l-Phe-CF3, N-Acetyl-l-Val-l-Phe-CF3, and N-Acetyl-l-Leu-l-Phe-CF3. The D/H fractionation factors (φ) for the hydrogen in the H-bond between His 57 and Asp 102 (His 57-Hδ1) in these four complexes at 5°C were in the range φ = 0.32–0.43, expected for a low-barrier hydrogen bond. For this series of complexes, measurements also were made of the chemical shifts of His 57-Hɛ1 (δ2,2-dimethylsilapentane-5-sulfonic acid 8.97–9.18), the exchange rate of the His 57-Hδ1 proton with bulk water protons (284–12.4 s−1), and the activation enthalpies for this hydrogen exchange (14.7–19.4 kcal⋅mol−1). It was found that the previously noted correlations between the inhibition constants (Ki 170–1.2 μM) and the chemical shifts of His 57-Hδ1 (δ2,2-dimethylsilapentane-5-sulfonic acid 18.61–18.95) for this series of peptidyl trifluoromethyl ketones with chymotrypsin [Lin, J., Cassidy, C. S. & Frey, P. A. (1998) Biochemistry 37, 11940–11948] could be extended to include the fractionation factors, hydrogen exchange rates, and hydrogen exchange activation enthalpies. The results support the proposal of low barrier hydrogen bond-facilitated general base catalysis in the addition of Ser 195 to the peptidyl carbonyl group of substrates in the mechanism of chymotrypsin-catalyzed peptide hydrolysis. Trends in the enthalpies for hydrogen exchange and the fractionation factors are consistent with a strong, double-minimum or single-well potential hydrogen bond in the strongest complexes. The lifetimes of His 57-Hδ1, which is solvent shielded in these complexes, track the strength of the hydrogen bond. Because these lifetimes are orders of magnitude shorter than those of the complexes themselves, the enzyme must have a pathway for hydrogen exchange at this site that is independent of dissociation of the complexes.
A low barrier hydrogen bond (LBHB) between His 57 and Asp 102 has been postulated to play an important role in the mechanism of action of chymotrypsin (1, 2). According to the LBHB-facilitated mechanism, the formation of an LBHB between His 57 and Asp 102 facilitates the formation of and stabilizes the tetrahedral intermediate formed by the addition of the 3-hydroxyl group of Ser 195 to the acyl carbonyl group of a substrate in the acylation step of catalysis (1). This mechanism is supported by the high basicity of His 57 in tetrahedral addition complexes of chymotrypsin with peptidyl trifluormethylketones (peptidyl-TFKs), which are analogs of tetrahedral intermediates, as illustrated in Scheme S1 (3, 4).
Scheme 1.

The mechanism is further supported by correlation of the pKas of these complexes with values of log Ki for the peptidyl-TFKs, by correlation of the pKas with values of log kcat/Km for substrates with the same peptidyl groups, and by correlation of the proton NMR chemical shifts of the LBHBs with pKa and log Ki for the peptidyl-TFKs (5).
The physicochemical properties of LBHBs include (i) very short contacts between the participating heavy atoms, (ii) very low-field proton chemical shifts, (iii) low fractionation factors, and (iv) positive values for the deuterium isotope effect on the NMR chemical shift of the LBHB (6, 7). In addition, LBHBs display high activation energies for exchange with medium protons (8, 9). Chymotrypsin or chymotrypsinogen in acidic solutions is, in one respect, analogous to the tetrahedral adduct between Ser 195 and a peptide carbonyl group, in that His 57 is protonated and an LBHB is postulated to exist between His 57 and Asp 102 (1, 3, 8). The assignment of an LBHB in the protonated diad His 57–Asp 102 of chymotrypsinogen was based on its low-field proton chemical shift (18.3 ppm), its low fractionation factor (0.4), and the moderately high activation energy (10.7 kcal⋅mol−1) for its exchange with the protons in water (8). Physicochemical evidence supporting the assignment of His 57-Hδ1 of chymotrypsin in peptidyl-TFK complexes to an LBHB include the change in the chemical shift of His 57-Hδ1 to higher frequency on complex formation (3–5) and the apparent close contact between His 57-Nδ1 and Asp 102-Oδ1 in crystal structures (10). In a related complex of a peptidyl-TFK with subtilisin, a D/H fractionation factor of φ = 0.85 was reported for His 57-Hδ1, which is consistent with its being a strong H-bond (12).
In the present paper, we describe experiments that reveal the D/H fractionation factors for His 57-Hδ1 in tetrahedral complexes between chymotrypsin and a series of four peptidyl-TFKs. We report the chemical shifts of the His 57-Hδ1 and His 57-Hɛ1 protons in the four complexes. We further report the lifetimes and activation energies for the exchange His 57-Hδ1 with solvent in three of these complexes. These data allow us to make correlations between the association constants for these complexes and spectral, kinetic, and thermodynamic parameters. The results provide the most complete analysis of LBHBs obtained to date for an enzyme system.
EXPERIMENTAL SECTION
Materials.
Chymotrypsin Aα was purchased from Sigma (type II, 3× crystallized from bovine pancreas). Peptidyl trifluoromethyl ketones were synthesized as described (13). The NMR experiments were performed on Bruker (Billerica, MA) DMX 750 and DMX 500 spectrometers. NMR spectra were processed with felix 2.3 software (MSI, San Diego).
Fractionation Factors.
The fractionation factors were determined at 5°C by integrating the low field proton signals in solutions of different H2O/D2O composition as described (8). The samples contained ≈2 mM chymotrypsin in 0.1 M phosphate buffer, and the pH was between 8.3 and 8.5. Peptidyl-TFKs were dissolved in dimethyl sulfoxide and were added to the chymotrypsin samples; the final dimethyl sulfoxide content was <4%. The levels of inhibitors added were 2 equivalents of N-acetyl-l-leucyl-l-phenylalanyl-TFK (N-AcLF-CF3), 1.7 equivalents of N-acetyl-l-valyl-l-phenylalanyl-TFK, 2 equivalents of N-acetyl-glycyl-l-phenylalanyl-TFK, and 2.5 equivalents of N-acetyl-l-phenylalanyl-TFK relative to chymotrypsin. A 1–1 pulse sequence was used to decrease the water signal with minimal saturation (14). Corrections in signal intensity with mole fraction H2O were made according to the procedure of Markley et al. (8). The fractionation factor (φ) was determined by fitting the data to Eq. 1:
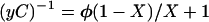 |
1 |
where y is the signal intensity, C is a constant and X is the mole fraction of H2O.
Chemical Shifts.
The samples were prepared as described above except that 2,2-dimethylsilapentane-5-sulfonic acid was added as an internal standard. The chemical shift of His 57-Hɛ1 was determined from a truncated driven nuclear Overhauser effect difference experiment (15) with irradiation at the position of the signal from His 57-Hδ1 for 25 ms.
Exchange Rates.
The exchange rate was determined from linewidth (Δv1/2) measurements over the temperature range 1–55°C. Linewidths were determined by fitting Lorentzian curves to the experimental spectra. The data were fitted to an Arrhenius plot according to Eqs. 2, 3, and 4 (9):
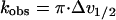 |
2 |
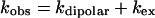 |
3 |
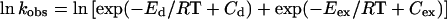 |
4 |
where Ed is the activation energy for the dipolar contribution and Eex is the activation energy for the exchange contribution. Cd and Cex are constants in the Arrhenius plot.
RESULTS
Fractionation factors of the low field protons in chymotrypsin-peptidyl-TFK complexes.
D/H fractionation factors were determined from the slopes of lines obtained in plots of the relative low-field signal intensities against mole fraction of H2O in H2O/D2O-mixtures according to Eq. 1, as illustrated for one case in Fig. 1. Their values for complexes of chymotrypsin with various peptidyl-TFKs with Phe in the P1 position and N-Ac, N-Ac-Gly, N-Ac-Val, and N-Ac-Leu in the P2 position are given in Table 1. The results show that the D/H fraction factor for the His 57-Hδ1 of chymotrypsin in each complex has a very low value (φ = 0.32–0.43) as compared with typical fractionation factors of 1.1 for N- and O-bonded protons in water (16).
Figure 1.

Determination of D/H fractionation factor. Shown is a representative plot to determine the fractionation factor for the low field proton in chymotrypsin and N-AcLF-CF3 complex. The relative intensity of the low field signal is plotted as a function of the mole fraction H2O (X) according to Eq. 1. The slope is the D/H fractionation factor.
Table 1.
Proton fractionation factors and chemical shifts of the diad His 57–Asp 102 in peptidyl-TFK complexes of chymotrypsin
| Inhibitor | Fractionation factor | His 57 chemical shift, ppm
|
Hydrogen exchange
|
Inhibition constants*Ki, μM | pKa of His 57-He2† | ||
|---|---|---|---|---|---|---|---|
| Hɛ1 | Hδ1† | kex, s−1§ | ΔH‡ (kcal⋅mol−1) | ||||
| N-AcF-CF3 | 0.32 ± 0.01 | 8.97 | 18.61 | 282 | 14.7 ± 4.5 | 17, 30 | 10.7 |
| 20, 40 | |||||||
| N-AcGF-CF3 | 0.34 ± 0.02 | 9.02 | 18.66 | 123 | 16.0 ± 3.7 | 18, 12 | 11.1 |
| N-AcVF-CF3 | 0.39 ± 0.02 | 9.09 | 18.91 | — | — | 2.8, 4.5 | 11.8 |
| N-AcLF-CF3 | 0.43 ± 0.20 | 9.18 | 18.95 | 12.4 | 19.4 ± 1.5 | 1.2, 2.4 | 12.1 |
Hydrogen Exchange with Solvent.
The temperature dependence of the width of the NMR signal from the His 57-Hδ1 of chymotrypsin in peptidyl-TFK complexes was found to be biphasic. On increasing the temperature from 0 to 55°C, the width of the signal at half-height first decreases and then increases. For the N-AcLF-CF3 complex, the transition occurs at ≈30°C (Fig. 2). The low field resonance is well defined at temperatures up to 55°C, indicating that the N-AcLF-CF3 is bound and the active site remains intact in the temperature range being studied. At low temperatures, the dipolar contribution to the width is predominant and results in a positive slope whereas at higher temperatures the contribution caused by exchange of the proton with solvent predominates and gives a negative slope.
Figure 2.

The exchange rate of the low field proton in the complex of N-AcLF-CF3 and chymotrypsin. Shown is an Arrhenius plot of the effect of temperature on the observed exchange rate of the low field resonance. The curve through the data is described by Eqs. 2, 3, and 4 by using Eex = 20.1 ± 1.1 kcal⋅mol−1, Cex = 36.4 ± 1.6, Ed −5.3 ± 0.2 kcal⋅mol−1, and Cd = −4.3 ± 0.4. The probe temperature was calibrated with neat methanol.
By fitting the data in Fig. 2 to Eq. 4, the rate constants for chemical exchange of the His 57-Hδ1 of chymotrypsin with water in three of the complexes were determined to be in the range 12.4–282 s−1 at 25°C (Table 1). The activation enthalpies for exchange varied between 14.7 and 19.4 kcal⋅mol−1 (Table 1).
Chemical Shifts of His 57-Hδ1 and His 57-Hɛ1.
For each chymotrypsin-peptidyl-TFK complex, the chemical shift of His 57-Hδ1 was determined from a 1–1 pulse sequence spectrum, and that of His 57-Hɛ1 was obtained from a truncated driven nuclear Overhauser effect difference spectrum as shown in Fig. 3. The chemical shifts (Table 1) clearly show both protons become increasingly deshielded with increasing affinity of the peptidyl-TFK for chymotrypsin.
Figure 3.

Determination of the chemical shift of Hɛ1 of His57 in chymotrypsin-peptidyl-TFK complexes. Shown are representative truncated driven nuclear Overhauser effect difference spectra of low field 1H NMR signals in the complex of N-AcLF-CF3 and chymotrypsin. The 18.95-ppm peak is assigned to the proton bridging His 57 and Asp 102. This signal was saturated selectively for 25 ms. The chemical shift of Hɛ1 is assigned as labeled. The peak labeled H57HN is from the nearby backbone amide proton of His 57 as described (8).
DISCUSSION
Fractionation Factors.
D/H fractionation factors have been used to characterize hydrogen bonds, with strong hydrogen bonds normally showing fractionation factors <1.0 (17). Low fractionation factors previously have been found in chymotrypsinogen (8) and subtilisin (12) at low pH values, in a complex of subtilisin with a peptidyl-TFK (12), and in Δ5-3-ketosteroid isomerase (18). Each of the hydrogens exhibiting such values has been postulated to be in a strong H-bond (or LBHB). The present results indicate that the hydrogen bond between His 57 and Asp 102 in peptidyl-TFK complexes of chymotrypsin is strong, which is consistent with the proposed mechanism of LBHB-facilitated general base catalysis in the action of chymotrypsin (1, 2, 4, 5, 8).
Plots of the fractionation factors (Table 1) against the association constants for the complexes, chemical shifts, or activation enthalpies for exchange yield relatively straight lines. The fractionation factors increase from 0.32 to 0.43 as the chemical shifts of the Hδ1 increase from 18.61 to 18.95 ppm and those of the Hɛ1 increase from 8.97 to 9.18 ppm.
Although the changes in φ are small and their uncertainties are relatively large, φ appears to increase from the very small value of 0.32 in the weakest complex as the strength of the hydrogen bond increases, as evidenced by the trends in the association constants, exchange rates, and exchange enthalpies. Such behavior is diagnostic for a LBHB as distinguished from a single-well hydrogen bond (1). As the N—O distance in such a hydrogen bond is shortened from the normal 2.8–2.9 Å, the fractionation factor decreases initially as the hydrogen becomes centrally located and less rigidly bonded to either heavy atom. A minimum in proton bonding is reached, however, beyond which further shortening of the N—O distance increases bonding and raises the fractionation factor. This occurs as the LBHB changes toward a more symmetrical single-well hydrogen bond (6). The trend in fractionation factors seen in Table 1 is an indication that the hydrogen bonds being observed are LBHBs in progression toward symmetrical, single-well hydrogen bonds. These data further indicate that the strength of an enzymatic LBHB can be varied in response to varying ligand structure. The change in fractionation factor in this series corresponds to the parallel change in the pKa of His 57 in this series of complexes (5).
Hydrogen Exchange with Solvent.
The rate constants for solvent exchange of the chymotrypsin His 57-Hδ1 in the TFK-complexes (Table 1) are much smaller than that for exchange of the low-field proton in chymotrypsinogen at low pH (952 s−1 at 1°C) (8) and ketosteroid isomerase (1,330 s−1 at 25°C) (18), suggesting that the LBHB-proton in the chymotrypsin-peptidyl-TFK complex is more strongly bound. The exchange rates for the LBHB protons in the chymotrypsin-peptidyl-TFK complexes are orders of magnitude faster than the dissociation rate constants for the peptidyl-TFKs in Table 1, which range from 3 × 10−4 to 3 × 10−2 s−1 (11). Inasmuch as the exchange rate constants in Table 1 range from 12.4 to 282 s−1, the exchange mechanism cannot include the dissociation of the peptidyl-TFKs. The chymotrypsin-peptidyl-TFK complexes are long-lived, so that a proton exchange mechanism that is independent of inhibitor dissociation accounts for the exchange rates. Whatever the mechanism of exchange, the rate is inversely related to the strength of the LBHB (Table 1).
The activation energy for the exchange of conventional acidic protons with water is generally low, in the range of 1–3 kcal⋅mol−1. However, the strongly bonded protons in LBHBs exhibit much higher activation energies. In the case of chymotrypsinogen at pH 3.5, the LBHB-proton bridging His 57 and Asp 102 undergoes exchange with an activation enthalpy of 10.7 kcal⋅mol−1 (8), and the activation enthalpy for the LBHB associated with Δ5-3-ketosteroid isomerase is 12.3 kcal⋅mol−1 (18). In the present work, the activation barrier to solvent exchange (ΔH‡) for each of the complexes is found to be in the range 14.7–19.4 kcal⋅mol−1 (Table 1). These values are larger than those for chymotrypsinogen at low pH and for ketosteroid isomerase, and they suggest that the LBHBs formed in N-AcLF-CF3 complexes of chymotrypsin are less accessible to solvent exchange and are very strong, perhaps the strongest LBHBs so far known in an enzyme. The results also show that the exchange is enthalpy driven by an activation enthalpy of 19.4 kcal⋅mol−1 and a free energy of 8.4 kcal⋅mol−1. An excellent correlation is observed between the inhibition constants for the peptidyl-TFKs and the activation enthalpies for exchange (Fig. 4).
Figure 4.

Correlation of log Ki for peptidyl-TFKs with enthalpies for LBHB exchange in chymotrypsin-peptidyl-TFK complexes. Shown is a plot of log Ki against the enthalpy of activation for exchange of the LBHB protons with water protons. The correlation coefficient is 0.905. Data are taken from Table 1.
Chemical Shifts of His 57-Hδ1 and His 57-Hɛ1.
The chemical shifts of these signals are indicative of a positively charged imidazole in the complex. For reference, the chemical shift of the protonated form of His 57 Hδ1 in free bovine chymotrypsin Aδ is 17.5 ppm (19), and that of free bovine chymotrypsinogen A is 18.1 ppm (8, 19). The chemical shift of protonated His 57-Hɛ1 is 8.50 ppm in chymotrypsin and 9.21 ppm in chymotrypsinogen (20). Thus, the values in the TFK-complexes (Table 1) are closer to those of the zymogen than the enzyme. As discussed above, the trend in the D/H fractionation factors indicates a shortening of both the Asp 102-Oδ1–His 57-Nδ1 and His 57-Hδ1–Asp 102-Oδ1 interatomic distances with increasing chemical shifts over the series of four TFK-complexes. This could have the effect of increasing the aromaticity of the imidazole ring, which would account for the concomitant deshielding of Hδ1 and Hɛ1. The observed effect is 50% larger on Hδ1 than on Hɛ1; this is in the same direction but much smaller than the 340% difference expected on the basis of observed protonation shifts of these signals in the uninhibited enzyme: 0.7 ppm for Hɛ1 and 2.4 ppm for Hδ1 (19, 20). Thus, explanation of the shifts requires either an additional effect that shields Hδ1 (for example, from the decrease in the Asp 102-Oδ1–His 57-Hδ1 distance, which might decrease the chemical shift toward that of a protonated carboxylate proton) or an additional effect that deshields Hɛ1 (as discussed below).
In explaining the chemical shift of His Hɛ1, the close contact between Hɛ1 and the carbonyl oxygen of Ser 214 must be taken into consideration. A study of all structurally characterized serine hydrolases containing an Asp(Glu)-His -Ser catalytic triad at their active centers shows that the Hɛ1 atom of the active site histidine is in van der Waals contact with a carbonyl oxygen (21). In chymotrypsin Aγ and acyl-chymotrypsin (22, 23), the distance between His 57-Cɛ1 and the carbonyl oxygen of Ser 214 is 3.04 Å. In chymotrypsinogen the distance is 3.13 Å.
However, a current software package that calculates proton shifts from structures (24) failed, in our hands, to reproduce the chemical shift difference between the Hɛ1 of chymotrypsin and chymotrypsinogen when given the x-ray structures of these proteins as input: the calculated shift change was in the wrong direction. Given limitations in the current level of understanding of proton chemical shifts, the exact mechanisms for these changes cannot be determined at present.
LBHB-Facilitated General Base Catalysis by His 57.
The decreased shielding of Hɛ1 and close contact between Cɛ1 and the carbonyl oxygen of Ser 214 are consistent with and support the LBHB-facilitated chymotrypsin mechanism (4, 5), which is illustrated in Fig. 5. Substrate binding is proposed to induce a conformational change leading to steric compression between His 57-Nδ1 and Asp 102-Oδ1. Strain in the compressed state can be relieved by the formation of an LBHB between His 57-Nδ1 and Asp 102δ1 because, in an LBHB, the heteroatoms are intrinsically close, i.e., compressed (6, 17). However, because the ΔpKa for the donor and acceptor atoms must be near zero in an LBHB, an LBHB cannot exist between His 57-Nδ1 and Asp 102-Oδ1 until a proton is added to His 57-Nɛ2. Compression, therefore, can lead to increased basicity of His 57 through LBHB-formation (4, 5). The ideal base strength for histidine would correspond to a pKa between 9 and 13, which are the approximate pKas for the leaving N-terminal amino group of the cleaved peptide and the nucleophilic Ser 195. The values of pKa for His 57 in the tetrahedral adducts of chymotrypsin with peptidyl-TFKs range from 10.3 to 12.1 (4, 5), about the middle of the optimal range.
Figure 5.

The postulated LBHB-facilitated mechanism for the acylation of chymotrypsin by a peptide substrate.
Because neither the imidazole of His 57 nor the carboxyl group of Asp 102 are in direct contact with the substrate, compression between them can take place only if they are forced together through the movement of other protein groups with which they maintain close contacts. Movement of the other groups induced by substrate binding could lead to compression between His 57-Nδ1 and Asp 102-Oδ1. One of these contacts may be that between Hɛ1 of His 57 and the carbonyl oxygen of Ser 214. Because they are in van der Waals contact, any substrate-induced translational movement of Ser 214 against the His 57-imidazole group could facilitate compression between His 57 and Asp 102. Such a role for Ser 214 could be one of several structural factors leading to compression; others would be required, for example, to hold the β-carboxyl group of Asp 102 in position. Alternatively, a compressing force on Asp 102 might be brought into play by substrate binding, with the resistance being provided by the carbonyl group of Ser 214 in contact with Cɛ1 of His 57. The trend in the chemical shift of Hɛ1 determined in the current study supports the concept that the binding of substrate or a substrate analog increases crowding and compression in the active center.
Based on the available data, it appears that the increasing size or hydrophobicity of the P2 residues in the series of peptidyl-TFKs studied here is correlated with increasing compression between His 57-Nδ1 and Asp 102-Oδ1, as well as with increasing strength of the LBHB (5). In related research, the size and hydrophobicity of the P2 residue is also correlated with increasing reactivity of the corresponding methyl esters as substrates (11).
Scheme 2.

Acknowledgments
This research was supported by Grant GM 51806 from the National Institute of General Medical Sciences. This study made use of the National Magnetic Resonance Facility at Madison, WI, which is supported by National Institutes of Health Grant RR02301 from the Biomedical Research Technology Program, National Center for Research Resources. Equipment in the facility was purchased with funds from the University of Wisconsin, the National Science Foundation Biological Instrumentation Program (Grant DMB-8415048), National Institutes of Health Biomedical Research Technology Program (Grant RR02301), National Institutes of Health Shared Instrumentation Program (Grant RR02781), and the U.S. Department of Agriculture.
ABBREVIATIONS
- LBHB
low barrier hydrogen bond
- TFK
trifluoromethyl ketone
- N-AcLF-CF3
N-acetyl-l-leucyl-l-phenylalanyl-TFK
References
- 1.Frey P A, Whitt S A, Tobin J B. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 2.Golubev N S, Denisov G S, Gindin V A, Ligay S S, Limbach H H, Smirnov S N. J Mol Struct. 1994;322:83–90. [Google Scholar]
- 3.Liang T-C, Abeles R H. Biochemistry. 1987;26:7603–7608. doi: 10.1021/bi00398a011. [DOI] [PubMed] [Google Scholar]
- 4.Cassidy C S, Lin J, Frey P A. Biochemistry. 1997;36:4576–4584. doi: 10.1021/bi962013o. [DOI] [PubMed] [Google Scholar]
- 5.Lin J, Cassidy C S, Frey P A. Biochemistry. 1998;37:11940–11948. doi: 10.1021/bi980278s. [DOI] [PubMed] [Google Scholar]
- 6.Hibbert F, Emsley J. Adv Phys Org Chem. 1990;26:255–379. [Google Scholar]
- 7.Jeffrey G A. An Introduction to Hydrogen Bonding. New York: Oxford Univ. Press; 1997. [Google Scholar]
- 8.Markley J L, Westler W M. Biochemistry. 1996;35:11092–11097. doi: 10.1021/bi961366k. [DOI] [PubMed] [Google Scholar]
- 9.Harris T K, Abeygunawardana C, Mildvan A S. Biochemistry. 1997;36:14661–14675. doi: 10.1021/bi972039v. [DOI] [PubMed] [Google Scholar]
- 10.Brady K, Wei A, Ringe D, Abeles R H. Biochemistry. 1990;29:7600–7607. doi: 10.1021/bi00485a009. [DOI] [PubMed] [Google Scholar]
- 11.Brady K, Abeles R H. Biochemistry. 1990;29:7608–7617. doi: 10.1021/bi00485a010. [DOI] [PubMed] [Google Scholar]
- 12.Halkides C J, Wu Y Q, Murray C J. Biochemistry. 1996;35:15941–15948. doi: 10.1021/bi961805f. [DOI] [PubMed] [Google Scholar]
- 13.Imperiali B, Abeles R H. Biochemistry. 1986;25:3760–3767. doi: 10.1021/bi00361a005. [DOI] [PubMed] [Google Scholar]
- 14.Hore P J. J Magn Reson. 1983;55:283–300. [Google Scholar]
- 15.Wagner G, Wüthrich K. J Magn Reson. 1979;33:675–680. [Google Scholar]
- 16.Quinn D M, Sutton L D. In: Enzyme Mechanism from Isotope Effects. Cook P F, editor. Boca Raton, Florida: CRC; 1991. pp. 73–126. [Google Scholar]
- 17.Kreevoy M M, Liang T M. J Am Chem Soc. 1980;102:3315–3322. [Google Scholar]
- 18.Zhao Q, Abeygunawardana C, Gittis A G, Mildvan A S. Biochemistry. 1997;36:14616–14626. doi: 10.1021/bi971549m. [DOI] [PubMed] [Google Scholar]
- 19.Robillard G, Shulman R G. J Mol Biol. 1974;76:541–558. doi: 10.1016/0022-2836(74)90179-x. [DOI] [PubMed] [Google Scholar]
- 20.Markley J L, Ibañez I B. Biochemistry. 1978;17:4627–4640. doi: 10.1021/bi00615a008. [DOI] [PubMed] [Google Scholar]
- 21.Derewenda Z S, Derewenda U, Kobos P M. J Mol Biol. 1994;241:83–93. doi: 10.1006/jmbi.1994.1475. [DOI] [PubMed] [Google Scholar]
- 22.Dixon M M, Brennan R G, Matthews B W. Int J Biol Macromol. 1991;13:89–96. doi: 10.1016/0141-8130(91)90054-x. [DOI] [PubMed] [Google Scholar]
- 23.Dixon M M, Matthews B W. Biochemistry. 1989;28:7033–7038. doi: 10.1021/bi00443a038. [DOI] [PubMed] [Google Scholar]
- 24.Williamson M P, Asakura T. J Magn Reson Ser B. 1993;10:63–71. [Google Scholar]