

Abstract
Background
Leishmania parasites cause a diverse spectrum of diseases in humans ranging from spontaneously healing skin lesions (e.g., L. major) to life-threatening visceral diseases (e.g., L. infantum). The high conservation in gene content and genome organization between Leishmania major and Leishmania infantum contrasts their distinct pathophysiologies, suggesting that highly regulated hierarchical and temporal changes in gene expression may be involved.
Results
We used a multispecies DNA oligonucleotide microarray to compare whole-genome expression patterns of promastigote (sandfly vector) and amastigote (mammalian macrophages) developmental stages between L. major and L. infantum. Seven per cent of the total L. infantum genome and 9.3% of the L. major genome were differentially expressed at the RNA level throughout development. The main variations were found in genes involved in metabolism, cellular organization and biogenesis, transport and genes encoding unknown function. Remarkably, this comparative global interspecies analysis demonstrated that only 10–12% of the differentially expressed genes were common to L. major and L. infantum. Differentially expressed genes are randomly distributed across chromosomes further supporting a posttranscriptional control, which is likely to involve a variety of 3'UTR elements.
Conclusion
This study highlighted substantial differences in gene expression patterns between L. major and L. infantum. These important species-specific differences in stage-regulated gene expression may contribute to the disease tropism that distinguishes L. major from L. infantum.
Background
Leishmania are protozoan parasites that cause a wide spectrum of clinical manifestations in humans, collectively referred to as leishmaniasis, ranging from self-resolving skin lesions (L. major and L. mexicana) to life-threatening visceral diseases (L. donovani and L. infantum) [1]. Leishmaniasis is endemic in 88 countries and the World Health Organization has estimated that more than 12 million people are currently infected with Leishmania and 350 million people are at risk of infection in tropical and subtropical regions of the world [1,2]. Leishmania parasites exist in two major developmental stages. In the alimentary tract of the sandfly vector, the parasites grow as extracellular flagellated promastigotes that are exposed to neutral pH and fluctuating temperatures averaging 25°C. Following the sand fly bite, the infective forms (metacyclic promastigotes) can be transferred to tissue macrophages of the mammalian host where they experience near-constant temperatures ranging from 35°C to 39°C and differentiate into aflagellated replicative amastigotes within the acidic phagolysosomal vacuoles [3]. During promastigote-to-amastigote differentiation, the parasites are subjected to drastic environmental changes, including a sharp rise in temperature, a drop in extracellular pH, an increased exposure to oxygen and nitrogen-reactive species, an intense extracellular proteolytic activity, and nutritional starvation. Several of these environmental signals trigger Leishmania differentiation [4-6] by activating many regulatory mechanisms affecting gene expression that result in important morphological and biochemical changes [7-10]. To date, several amastigote-specific [11-15] and promastigote-specific [16-18] genes have been identified in Leishmania, however, the molecular mechanisms governing developmental gene regulation in this organism warrant additional investigations.
Leishmania and the related Trypanosoma species possess unusual mechanisms of gene expression. The recent completion of the Leishmania spp. genomes indicates that protein-coding genes are organized as large polycistronic units [19,20]. Transcription has been postulated to initiate at strand switch regions on each chromosome [21] in the absence of defined RNA pol II promoters and typical general transcription factors. The maturation of individual mRNAs from polycistronic pre-mRNAs requires posttranscriptional control, which involves two coupled co-transcriptional RNA-processing reactions. These include trans-splicing where a capped RNA of ~39-nucleotides, the spliced leader RNA, is added to the 5'-terminus of all known protein-encoding RNAs, and 3'-end cleavage and polyadenylation (reviewed in [22]). Developmental gene regulation in Leishmania is determined posttranscriptionally mainly by sequences located in the 3'-untranslated regions (3'UTR). Several distinct 3'UTR elements have been identified among stage-specific transcripts to regulate mRNA stability/degradation and mRNA translation (reviewed in [22,23]).
The recent completion of the L. major and L. infantum genomic sequences [24] allowed studies of global gene expression throughout developmental life stages of these parasites. Global gene expression profiling using Leishmania microarrays with genome coverage between 22% and 97.5% highlighted that 2–9% of all genes analyzed were developmentally regulated [25-29]. The current study extends these microarray data by providing a detailed analysis of whole-genome stage- and species-specific gene expression profiles within L. major and L. infantum using a DNA oligonucleotide microarray representing the entire genomes of these two species. None of the previous DNA microarray studies has compared global gene expression profiling of promastigote (extracellular) versus amastigote (intracellular) forms of L. infantum, the causative agent of visceral leishmaniasis, the most severe form of the disease. Moreover, no studies to date have compared global changes in mRNA abundance during development between Leishmania species associated with different disease tropism (e.g. cutaneous vs. visceral leishmaniasis). The current comparative analyses revealed important differences in stage-regulated gene expression patterns between L. major and L. infantum. These species-specific differences may partly explain the distinct clinical pathologies, despite highly conserved genomes.
Results
Comparison of global gene expression profiles between promastigote and amastigote developmental stages of Leishmania spp
To investigate global mRNA expression profiles of L. infantum and L. major promastigote and amastigote developmental life stages, we designed a high-density multispecies 70-mer oligonucleotide genome microarray representing the entire genomes of L. major and L. infantum that share over 99% of their genes and contain species-specific genes. This allows a comparative analysis under the same conditions. Through a rigorous statistical approach, data from several independent L. infantum and L. major DNA microarray experiments were compiled and compared here. After subtracting the background, the difference of 1.7-fold in the signal intensity between the experimental conditions used (promastigote vs. amastigote RNA) for a given gene was chosen as the cut-off given that the p value confidence was more than 95% under those conditions. The whole-genome expression patterns of L. infantum (Figure 1A) and L. major (Fig. 1B) developmental stages are shown in the scatter plots of normalized data. The scatter plots compared each gene according to the normalized log2 ratio of the Alexa 647/Alexa 555 signal intensities (amastigotes/promastigotes) and to the signal mean intensity of each spot. More than 86% of the spots yielded hybridization intensities of two-fold over local background. Non-modulated genes, considered as constitutively expressed, had ratios between 0.6 and 1.7 (log2 = ± 0.75). Remarkably, most Leishmania spots yielded expression ratios close to 1 (log2 = 0), with only a few hundred genes showing modulation of greater than 1.7-fold between developmental stages in both species (Figure 1 and Table 1). The vast majority of genes (91–93%) in both Leishmania species were not significantly modulated (less than 1.7-fold modulation at the level of mRNA expression) throughout the two life cycle stages studied.
Figure 1.

Whole-genome expression profiling of amastigote vs. promastigote upregulated transcripts in Leishmania spp. Scatter plots showing the distribution of signal intensities generated by the DNA microarray experiments employing total RNA of L. infantum (A) and L. major (B) extracted from promastigote (Pro) and amastigote (Ama) developmental life stages. The horizontal axis displays the normalized mean signal intensity for each gene (([Pro]+ [Ama]/2) and the vertical axis shows the normalized ratio of amastigote versus promastigote gene expression ([Ama]/[Pro]). External line represents a 1.7-fold change. Genes significantly upregulated in the amastigote stage are above the line at 1.7 and genes upregulated in the promastigote stage are below the line at 0.6. The data presented here are the average of six independent biological replicates for L. infantum and four independent biological replicates for L. major.
Table 1.
Patterns of global differential gene expression in Leishmania infantum and Leishmania majora.
| L. infantum | L. major | |||
| Fold increase | promastigotes | amastigotes | promastigotes | amastigotes |
| 1.7–3.0 | 260 | 217 | 413 | 233 |
| 3.1–6.0 | 14 | 52 | 63 | 35 |
| 6.1–9.0 | 0 | 19 | 4 | 15 |
| 9.1–12.0 | 0 | 9 | 1 | 7 |
| > 12.1 | 0 | 12 | 0 | 11 |
| Total | 274 | 309 | 481 | 301 |
| % of modulated genes | 3.3 | 3.8 | 5.7 | 3.6 |
a All genes presented here (total 1365) showed a fold increase in mRNA accumulation more than 1.7 and a p value < 0.05.
Comparison of global gene expression profiles of Leishmania infantum developmental stages
To date, no studies on the global stage-specific gene expression of a visceralizing Leishmania species have been reported. We therefore undertook a microarray-based approach to determine differential gene expression patterns of the two major L. infantum life cycle stages: (i) non-infective replicating extracellular procyclic promastigotes, and (ii) intracellular amastigotes isolated from THP-1-infected human monocytes in vitro following 4 days post-infection. Hybridizations were carried out using RNA from six experimental biological replicates. After normalization and data processing in order to assess differential gene expression, the data were first filtered by FDR-corrected p-value (p < 0.05) and then according to the modulation of each probe (see Methods). This analysis led to 583 genes (7.3% of the total L. infantum genome) showing > 1.7-fold change in mRNA abundance between the promastigote and amastigote life stages (Table 1). Analysis of the stage-modulated genes according to their gene ontology (GO) showed that they belong to several categories, including various biological processes (see Additional files 1 and 2 and Figure 2A–B).
Figure 2.

Distribution of Leishmania infantum and Leishmania major differentially expressed genes according to Gene Ontology (GO) function categories. GO categories for L. infantum promastigotes (A), L. infantum amastigotes (B), L. major promastigotes (C) and L. major amastigotes (D). The majority of genes encode unknown biological functions. Unclassified proteins include the hypothetical proteins (proteins with unknown function and not tested experimentally) and proteins with no GO category (unclassified) that have been experimentally characterized (e.g. amastins).
Two hundred and seventy four genes representing 3.3% of the L. infantum genome were preferentially expressed in procyclic promastigotes (listed in Additional file 1). Fold-increase in mRNA abundance was modest (1.7- to 3-fold) for 95% of the overexpressed transcripts (Table 1). This group included the well-documented promastigote-regulated genes PFR1D and PFR2C [18], several dyneins and kinesins that are important for flagellar movement, cytokinesis, and motility [30,31], histones [32,33], kinetoplast DNA-associated proteins, the U-rich RNA-binding proteins UPB1 and UBP2 [34] and a member of the Pumilio family of translation repressors [35], calpain cysteine peptidases and several proteasome subunits (see Additional file 1). Half of the promastigote-upregulated genes (49%) encoded hypothetical proteins with no similarity to other known proteins in the databases.
Three hundred and nine genes, representing 3.8% of the total L. infantum protein-coding sequences (Table 1) were found upregulated in intracellular amastigotes compared to extracellular promastigotes (listed in Additional file 2). Transcripts modulated specifically in amastigotes showed generally a higher accumulation than promastigote-upregulated transcripts, with 31% of the transcripts demonstrating more than 3-fold increase in expression levels (average of 9.5-fold) (Table 1). Several well-documented gene families, including the amastins [15], members of the glycosylation phosphoglycan beta 1,3 galactosyltransferase gene family (SCG: Side Chain Galactose) [36] and SHERP1, a small hydrophilic protein localizing to the endoplasmic reticulum and outer mitochondrial membrane [29,37] were upregulated in L. infantum amastigotes. Other representative examples included the myo-inositol-1-phosphate synthase, a mRNA capping methyltransferase involved in cap4 methylation of the spliced-leader RNA [38], aminopeptidases and the hs1vu complex proteolytic subunit and several ABC, amino acid and mitochondrial transporters (see Additional file 2). Approximately 74% of the amastigote-upregulated genes encoded hypothetical or unclassified proteins for which no putative biological function has been assigned.
Comparison of global gene expression profiles of different developmental stages within Leishmania major
In order to compare mRNA expression profiles between L. infantum and L. major developmental life stages, we undertook also DNA microarray experiments with L. major. In this study, we used the L. major LV39 MRHO/SU/59/P strain instead of the L. major Friedlin genome strain (MHOM/IL/81/Friedlin) because in our hands L. major LV39 was more infectious in BALB/c mice. Hybridizations were performed using RNA from four experimental biological replicates. Using the same statistical analysis applied for L. infantum, we identified 782 differentially expressed transcripts in L. major (9.3% of the total L. major genes) from which 481 were preferentially expressed in procyclic promastigotes (5.7%) and 301 were differentially expressed in lesion-derived amastigotes (3.6%) (Table 1). Recent microarray analyses reported that 1.5% of the L. major genes were differentially expressed in promastigotes and 1.4% in lesion-derived amastigotes [28]. The observed differences between the two studies may be attributed to the different L. major strains used or to technical issues regarding RNA preparations from mice lesions and/or to differences in the oligonucleotide genome microarray design and manipulation. The 782 identified differentially expressed genes were grouped according to GO functional categories (see Additional files 3 and 4 and Figure 2C–D). The vast majority of the transcripts enriched in L. major promastigotes (86%) showed a modest accumulation not exceeding 3-fold. Similarly to L. infantum, several L. major amastigote-upregulated transcripts showed in average a higher differential accumulation than the promastigote-upregulated transcripts (Table 1). Representative genes among those specifically upregulated in L. major promastigotes encode sugar kinases and beta-fructofuranosidases involved in carbohydrate metabolism, fatty acid elongases involved in lipid metabolism, proteins participating in nucleoside-nucleotide metabolism, amino acid, glucose and pteridine transport, electron transport, proteolysis (e.g. members of the calpain-like cysteine peptidase and peptidyl dipeptidase families), signaling (e.g. MAP and serine/threonine kinases, a PP1 Ser/Thr phosphatase, calmodulins, receptor-type adenylate cyclase a and b) and a large number of microtubule-associated proteins (see Additional file 3). This group also included several well-documented differentially expressed genes in promastigotes such as histones [26,39], the glucose transporter GT2 [14], the paraflagellar rod protein PFR1D and PFR2C [18], the leishmanolysin GP63 (also called MSP) [40,41], and the surface antigen protein 2 gene family (also known as GP46) [16,42]. More than 47% of the promastigote-upregulated transcripts encoded hypothetical proteins of unknown function (see Additional file 3). Among the L. major amastigote-upregulated genes are included several well-documented gene families such as amastins [15], HASPA1,2 [37], SCG5 and SCG7 which are members of the phosphoglycan beta 1,3 galactosyltransferase gene family [36], and others such as RNA-binding proteins, cathepsin L-like proteins, protein kinases, tryparedoxins, a family of novel antioxidant proteins, several amino acid and pteridine transporters and a large number (51%) of hypothetical proteins (see Additional file 4).
Comparison of global stage-regulated gene expression between L. infantum and L. major
One of the goals of this study was to compare global gene expression within the same life cycle stages of the L. major and L. infantum species, the causative agents of cutaneous and visceral leishmaniasis, respectively. Sequence comparison between the L. major and L. infantum genomes revealed marked conservation of synteny [20]. Therefore, much of the species-specific adaptive strategies to different target tissues and distinct disease pathogenesis should depend on the non-syntenic part of their respective genomes and/or on highly regulated hierarchical and temporal changes in gene expression. To monitor global gene expression of L. infantum and L. major, we used a multispecies high-density oligonucleotide microarray. It has recently been reported that using a multispecies microarray to study related species provided superior inter-species comparison than using several single species-specific microarrays [43]. Our comparative microarray analyses showed that out of the 755 genes differentially expressed in L. infantum and L. major promastigote forms (Figure 3) only 91 (12.05%) were in common between the two species. Similarly, only 64 (10.5%) of the 610 amastigote-upregulated genes were common to both species. These results are displayed in the form of Venn diagrams (Figure 3) and the detailed list of the commonly upregulated genes between L. infantum and L. major for a given developmental life stage is provided in Table 2. This list included, among others, the amastigote-specific amastin surface proteins, the paraflagellar rod components upregulated in promastigotes, dyneins, members of the phosphoglycan beta 1,3 galactosyltransferase, calpain-like cysteine peptidases, calmodulins and a large number of hypothetical proteins.
Figure 3.

Comparison of differential mRNA expression between Leishmania major and Leishmania infantum. Venn diagram comparing L. infantum and L. major genes significantly upregulated (> 1.7-fold, p < 0.05) in either log-phase promastigotes or intramacrophage amastigotes. The intersection of the Venn diagrams shows the number of genes that were common to both species. These common genes are listed in Table 2. The numbers within separate circles correspond to differentially expressed genes that are unique to each species. Species comparison was performed only on probes that had less than two mismatches when hybridized to either Leishmania species. Thus, ~7000 probes could be directly compared between the two organisms.
Table 2.
Differentially expressed genes common to Leishmania infantum and Leishmania major.
| GO annotation (molecular function) | Accession number (GeneDB) L. infantum | Fold increase | Accession number (GeneDB L. major | Fold increase |
| Metabolic process | ||||
| 3-ketoacyl-coa thiolase | LinJ23_V3.0860 | 2,1 a | LmjF23.0690 | 3,3 a |
| hydrolase, alpha/beta fold family | LinJ17_V3.1110 | 1,7 a | LmjF17.1010 | 1,8 a |
| vacuolar ATP synthase subunit B | LinJ28_V3.2610 | 1,8 a | LmjF28.2430 | 1,7 a |
| succinyl-coA:3-ketoacid-coenzyme A transferase b | LinJ33_V3.2470 | 1,8 c | LmjF33.2340 | 2,1 a |
| hydrolase, alpha/beta fold family | LinJ17_V3.1110 | 1,7 c | LmjF17.1010 | 1,8 c |
| Carbohydrate metabolic process | ||||
| acetyl-CoA synthetase | LinJ23_V3.0880 | 2,0 a | LmjF23.0710 | 4,5 a |
| aldose 1-epimerase | LinJ35_V3.1000 | 1,8 a | LmjF35.0980 | 2,4 a |
| enolase | LinJ14_V3.1240 | 2,3 a | LmjF14.1160 | 2,4 a |
| myo-inositol-1-phosphate synthetase | LinJ14_V3.1450 | 2,4 c | LmjF14.1360 | 4,7 c |
| Lipid metabolic process | ||||
| 3-oxo-5-alpha-steroid 4-dehydrogenase | LinJ25_V3.1850 | 1,7 a | LmjF25.1770 | 3,0 a |
| fatty acid elongase | LinJ14_V3.0700 | 2,0 a | LmjF14.0670 | 3,2 a |
| lathosterol oxidase | LinJ23_V3.1560 | 1,9 a | LmjF23.1300 | 3,6 a |
| phosphoglycan beta 1,3 galactosyltransferase | LinJ02_V3.0140 | 1,8 a | LmjF02.0160 | 3,8 a |
| phosphoglycan beta 1,3 galactosyltransferase d | 22,0 c | 2,4-11,8 c | ||
| hypothetical | LinJ13_V3.0200 | 2,7 c | LmjF13.0200 | 3,8 c |
| Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | ||||
| adenylosuccinate synthetase | LinJ13_V3.1090 | 1,8 a | LmjF13.1190 | 2,0 a |
| LinJ25_V3.1210 | ||||
| LinJ25_V3.2580 | LmjF25.1170, | |||
| ATPase beta subunit | LinJ25_V3.2590 | 2,1 a | LmjF25.1180 | 1,9 a |
| DNA metabolic process | ||||
| 3'-nucleotidase/nuclease | LinJ12_V3.0350 | 2,0 a | LmjF12.0400 | 3,9 a |
| 3'-nucleotidase/nuclease precursor | LinJ31_V3.2380 | 1,7 a | LmjF31.2310 | 4,6 a |
| histone 1 | LinJ33_V3.3390 | 2,6 a | LmjF33.3240 | 1,9 a |
| 3'-nucleotidase/nuclease | LinJ31_V3.2370 | 2,0 c | LmjF31.2300 | 1,7 c |
| double-strand-break repair protein rad21 | LinJ05_V3.1090 | 3,0 c | LmjF05.1090 | 1,9 c |
| RNA metabolic process | ||||
| exosome complex exonuclease RRP45 | LinJ22_V3.1430 | 3,1 c | LmjF22.1580 | 1,8 c |
| RNA-binding protein 5 | LinJ09_V3.0080 | 3,4 c | LmjF09.0060 | 1,7 c |
| Amino acid and derivative metabolic process | ||||
| glutamate dehydrogenase | LinJ15_V3.1070 | 1,8 a | LmjF15.1010 | 1,8 a |
| Protein metabolic process | ||||
| HSP 70 | LinJ32_V3.2050 | 2,0 a | LmjF32.1940 | 1,9 a |
| Proteolysis | ||||
| LinJ14_V3.0910, | ||||
| calpain-like cysteine peptidase | LinJ14_V3.0920 | 1,7 a | LmjF14.0850 | 4,5 a |
| calpain-like cysteine peptidase | LinJ20_V3.1320 | 2,0 a | LmjF20.1280 | 2,1 a |
| calpain-like cysteine peptidase | LinJ20_V3.1350 | 1,7 a | LmjF20.1310 | 2,9 a |
| calpain-like cysteine peptidase | LinJ27_V3.2490 | 1,8 a | LmjF27.0510 | 2,8 a |
| calpain-like cysteine peptidase | LinJ32_V3.1020 | 1,8 a | LmjF32.0970 | 1,9 a |
| LmjF01.0830, | ||||
| LmjF02.0740, | ||||
| peptidyl-dipeptidase | LinJ02_V3.0710 | 1,7 a | LmjF27.2660 | 2,5 a |
| puromycin-sensitive aminopeptidase-like | LinJ12_V3.0830 | 2,1 c | LmjF12.1250 | 1,8 c |
| Protein modification process | ||||
| protein kinase A catalytic subunit | LinJ18_V3.1090 | 1,9 a | LmjF18.1080 | 3,2 a |
| protein kinase A regulatory subunit | LinJ13_V3.0160 | 2,5 a | LmjF13.0160 | 2,1 a |
| protein kinase | LinJ30_V3.1780 | 2,5 c | LmjF30.1780 | 1,7 c |
| Electron transport | ||||
| trypanothione synthetase | LinJ23_V3.0500 | 1,7 c | LmjF23.0460 | 1,8 c |
| Cellular component organization and biogenesis | ||||
| dynein heavy chain | LinJ13_V3.1390 | 1,7 a | LmjF13.1650 | 2,1 a |
| dynein heavy chain | LinJ26_V3.1000 | 1,9 a | LmjF26.1020 | 1,7 a |
| dynein heavy chain | LinJ28_V3.0650 | 1,8 a | LmjF28.0610 | 2,0 a |
| dynein heavy chain (pseudogene) | LinJ27_V3.2460 | 2,1 a | LmjF27.2590 | 2,0 a |
| dynein light chain | LinJ24_V3.1050 | 2,0 a | LmjF24.1030 | 2,0 a |
| dynein-associated roadblock | LinJ35_V3.1740 | 1,8 a | LmjF35.1750 | 2,1 a |
| OSM3-like kinesin | LinJ17_V3.0890 | 1,7 a | LmjF17.0800 | 2,0 a |
| Cell motility | ||||
| LmjF39.1750, | ||||
| LinJ29_V3.1880, | LmjF29.1760 | |||
| PFR 1D | LinJ29_V3.1890 | 3,9 a | LmjF29.1770 | 4,5 a |
| LinJ16_V3.1510, | LmjF16.1425, | |||
| PFR 2C | LinJ16_V3.1520 | 3,7 a | LmjF16.1430 | 5,3 a |
| paraflagellar rod component | LinJ09_V3.1390 | 1,9 a | LmjF09.1320 | 2,8 a |
| Cell communication | ||||
| LmjF09.0910, | ||||
| LinJ09_V3.0970, | LmjF09.0920 | |||
| calmodulin | LinJ09_V3.0980 | 2,0 a | LmjF09.0930 | 2,8 a |
| calmodulin | LinJ13_V3.1060 | 1,8 a | LmjF13.1160 | 1,8 a |
| receptor-type adenylate cyclase a | LinJ17_V3.0120 | 1,7 a | LmjF17.0200 | 1,8 a |
| LmjF17.0230, | ||||
| LinJ17_V3.0140, | LmjF17.0235 | |||
| receptor-type adenylate cyclase b | LinJ17_V3.0160 | 1,8 a | LmjF17.0237 | 2,6 a |
| Transport | ||||
| amino acid transporter aATP11 b | LinJ31_V3.0370 | 2,4 c | LmjF31.0350 | 4,7 a |
| phosphate-repressible phosphate permease | LinJ03_V3.0480 | 1,7 c | LmjF03.0500 | 2,0 c |
| Unclassified | ||||
| oxidoreductase | LinJ36_V3.4380 | 1,7 a | LmjF36.4170 | 1,7 a |
| leucine rich repeat protein | LinJ32_V3.3200 | 1,7 a | LmjF32.3010 | 2,2 a |
| leucine rich repeat protein | LinJ10_V3.0160 | 2,1 a | LmjF10.0180 | 2,3 a |
| protein tyrosine phosphatase | LinJ05_V3.0280 | 1,8 a | LmjF05.0280 | 2,6 a |
| hypothetical | LinJ01_V3.0640 | 2,4 a | LmjF01.0620 | 2,3 a |
| hypothetical | LinJ02_V3.0520 | 3,9 a | LmjF02.0550 | 5,0 a |
| hypothetical | LinJ07_V3.0040 | 2,0 a | LmjF07.0030 | 1,8 a |
| hypothetical | LinJ07_V3.0470 | 1,8 a | LmjF07.0310 | 2,7 a |
| hypothetical | LinJ09_V3.1620 | 2,1 a | LmjF09.1530 | 1,8 a |
| hypothetical | LinJ10_V3.1370 | 1,9 a | LmjF10.1230 | 1,9 a |
| hypothetical | LinJ11_V3.0620 | 2,0 a | LmjF11.0610 | 2,1 a |
| hypothetical | LinJ11_V3.1040 | 2,1 a | LmjF11.1040 | 2,0 a |
| hypothetical | LinJ17_V3.0970 | 2,0 a | LmjF17.0870 | 3,6 a |
| hypothetical | LinJ18_V3.1640 | 3,5 a | LmjF18.1640 | 6,6 a |
| hypothetical | LinJ19_V3.0520 | 4,5 a | LmjF19.0520 | 5,9 a |
| hypothetical | LinJ20_V3.0760 | 1,7 a | LmjF20.0700 | 1,8 a |
| hypothetical | LinJ21_V3.0440 | 1,8 a | LmjF21.0380 | 2,5 a |
| hypothetical | LinJ23_V3.1190 | 2,0 a | LmjF23.1020 | 4,6 a |
| hypothetical | LinJ24_V3.1630 | 4,1 a | LmjF24.1560 | 4,1 a |
| hypothetical | LinJ24_V3.2200 | 2,4 a | LmjF24.2110 | 1,8 a |
| hypothetical | LinJ26_V3.2400 | 2,1 a | Lm jF26.2380 | 3,0 a |
| hypothetical | LinJ27_V3.0720 | 1,9 a | LmjF27.0870 | 1,7 a |
| hypothetical | LinJ28_V3.1150 | 2,1 a | LmjF28.1060 | 2,9 a |
| hypothetical | LinJ29_V3.0360 | 1,7 a | Lm jF29.0350 | 1,9 a |
| hypothetical | LinJ29_V3.1090 | 2,4 a | Lm jF29.1000 | 3,0 a |
| hypothetical | LinJ29_V3.1190 | 2,0 a | Lm jF29.1100 | 1,8 a |
| hypothetical | LinJ29_V3.1260 | 2,5 a | Lm jF29.1170 | 3,2 a |
| hypothetical | LinJ29_V3.2940 | 2,5 a | Lm jF29.2830 | 3,7 a |
| hypothetical | LinJ30_V3.2870 | 4,4 a | Lm jF30.2850 | 8,5 a |
| hypothetical | LinJ31_V3.1220 | 2,7 a | LmjF31.1200 | 2,0 a |
| hypothetical | LinJ32_V3.0360 | 1,7 a | Lm jF32.0350 | 2,0 a |
| hypothetical | LinJ32_V3.0370 | 2,2 a | Lm jF32.0360 | 2,2 a |
| hypothetical | LinJ32_V3.1840 | 2,2 a | Lm jF32.1760 | 3,0 a |
| hypothetical | LinJ32_V3.2020 | 2,8 a | Lm jF32.1910 | 2,4 a |
| hypothetical | LinJ33_V3.0660 | 3,7 a | Lm jF33.0610 | 5,4 a |
| hypothetical | LinJ34_V3.1620 | 2,0 a | Lm jF34.1520 | 4,0 a |
| hypothetical | LinJ34_V3.2590 | 1,7 a | LmjF34.2760 | 1,8 a |
| hypothetical | LinJ34_V3.4230 | 1,9 a | Lm jF34.4600 | 3,0 a |
| hypothetical | LinJ35_V3.5310 | 2,4 a | Lm jF35.5340 | 4,0 a |
| hypothetical | LinJ36_V3.0800 | 2,4 a | LmjF36.0740 | 1,8 a |
| hypothetical | LinJ36_V3.3000 | 1,8 a | Lm jF36.2850 | 2,0 a |
| hypothetical | LinJ36_V3.3780 | 1,8 a | Lm jF36.3620 | 6,7 a |
| hypothetical | LinJ36_V3.4440 | 4,1 a | Lm jF36.4230 | 5,7 a |
| hypothetical | LinJ36_V3.5010 | 2,7 a | Lm jF36.4780 | 3,2 a |
| hypothetical | LinJ36_V3.5140 | 2,3 a | LmjF36.4910 | 1,9 a |
| hypothetical | LinJ36_V3.5210 | 1,8 a | LmjF36.4980 | 2,5 a |
| hypothetical | LinJ35_V3.3780 | 1,8 c | LmjF35.3730 | 1,7 a |
| LmjF 23.1050, | ||||
| LinJ23_V3.1210, | LmjF 23.1080, | |||
| SHERP b | LinJ23_V3.1230 | 2,5 c | LmjF23.1086 | 1,7 a |
| Amastins e | 1.9–6.8 c | 1.9–25.4 c | ||
| LmjF 05.1230, | ||||
| LmjF19.1650 | ||||
| GIPL galf transferase | LinJ32_V3.4140 | 2,8 c | LmjF32.3990 | 2,0 c |
| hypothetical | LinJ01_V3.0650 | 1,7 c | LmjF01.0630 | 2,5 c |
| hypothetical | LinJ06_V3.1030 | 2,1 c | LmjF06.0995 | 1,8 c |
| hypothetical | LinJ08_V3.0650 | 2,0 c | Lm jF08.0640 | 4,8 c |
| hypothetical | LinJ10_V3.1130 | 2,7 c | LmjF10.1050 | 1,8 c |
| hypothetical | LinJ12_V3.0440 | 1,9 c | Lm jF12.0480 | 5,1 c |
| hypothetical | LinJ16_V3.0100 | 1,9 c | Lm jF16.0090 | 2,5 c |
| hypothetical | LinJ24_V3.2320 | 2,7 c | LmjF24.2230 | 1,7 c |
| hypothetical | LinJ25_V3.2870 | 2,3 c | LmjF35.2820 | 1,8 c |
| hypothetical | LinJ26_V3.1440 | 1,8 c | Lm jF26.1460 | 2,0 c |
| hypothetical | LinJ27_V3.0770 | 20,8 c | Lm jF27.0910 | 1,9 c |
| hypothetical | LinJ27_V3.2320 | 2,5 c | LmjF27.2370 | 1,8 c |
| hypothetical | LinJ30_V3.0820 | 2,0 c | LmjF30.0770 | 1,8 c |
| hypothetical | LinJ30_V3.1000 | 7,1 c | LmjF30.0940 | 2,7 c |
| hypothetical | LinJ30_V3.2340 | 1,7 c | Lm jF30.2330 | 2,4 c |
| hypothetical | LinJ31_V3.1190 | 2,1 c | Lm jF31.1170 | 2,7 c |
| hypothetical | LinJ 31_V 3.2140 | 1,9 c | LmjF 31.2090 | 2,0 c |
| hypothetical | LinJ 32_V3.3600 | 1,9 c | LmjF 32.3400 | 2,3 c |
| hypothetical | LinJ 33_V3.1720 | 4,2 c | Lm jF 33.1620 | 4,5 c |
| hypothetical | LinJ 35_V3.5130 | 1,7 c | Lm jF 35.5160 | 1,8 c |
a Genes differentially expressed in promastigotes.
b Genes differentially expressed but not in the same life stage.
c Genes differentially expressed in amastigotes.
d The majority of the phosphoglycan 1,3 galactosyltransferase gene family members are differentially expressed in amastigotes. These genes are listed in Additional files 2 and 4.
e The majority of the amastin gene family members are differentially expressed in amastigotes. These genes are listed in Additional files 2 and 4.
One hundred and nineteen genes were differentially expressed in L. infantum but were not modulated in L. major (Table 3). More than 50% of these genes encoded hypothetical proteins. For example, the LinJ29_V3.0930 gene was overexpressed 47-fold in L. infantum amastigotes but mRNA expression of its L. major orthologue remained unchanged. Other examples of genes upregulated in L. infantum amastigotes but not developmentally regulated in L. major included the aminopeptidase LinJ33_V3.2700 and metallopeptidases LinJ16_V3.0850 and LinJ34_V3.1130, the ABC transporters LinJ29_V3.0640 and LinJ11_V3.0040, the Rab GTPase activator protein LinJ29_V3.1670, the chaperone DNAJ LinJ21_V3.0550, the kinesin LinJ25_V3.2050, the mRNA capping methyltransferase LinJ36_V3.0130 and the nuclear cap binding protein LinJ30_V3.0560 (Table 3). Alternatively, 131 genes were stage-regulated in L. major but constitutively expressed in L. infantum (Table 4). Most of these L. major species-regulated genes were predominantly expressed in promastigotes. These included genes involved in carbohydrate metabolism like the three beta-fructofuranosidases in the L. major genome, 6-phospho-1-fructokinase LmjF29.2510 and hexokinase LmjF21.0250, genes participating in proteolysis such as the calpain-like cysteine peptidases LmjF20.1190 and LmjF27.0510 and carboxypeptidases LmjF13.0090 and LmjF33.2540, genes involved in electron transport such as the lactate dehydrogenase LmjF29.0280, the amino acid permease LmjF27.0680 and the transmembrane amino acid transporter LmjF07.1160 (9.7-fold of regulation), the glucose transporters lmgt2 LmjF36.6280/LmjF36.6290 and the surface antigens prostaglandin f2-alpha synthase and membrane-bound acid phosphatase 2 (Table 4). Previous studies have reported that the membrane-bound acid phosphatase 2 is a marker of virulence of L. donovani promastigotes [44] and that in L. mexicana this gene was not required for amastigote survival [45].
Table 3.
Genes differentially expressed in Leishmania infantum but constitutively expressed in Leishmania major.
| GO annotation (molecular function) | Accession number (GeneDB) | Fold increase | Accession number (GeneDB) | Fold increase a |
| L. infantum | L. major | |||
| Metabolic process | ||||
| glycerolphosphate mutase | LinJ33_V3.2220 | 2,2 b | LmjF33.2100 | 1,4 |
| iron superoxide dismutase | LinJ32_V3.1920 | 1,9 b | LmjF32.1839 | 1,2 |
| proteasome alpha 7 subunit | LinJ27_V3.0190 | 1,9 b | LmjF27.0190 | 1,3 |
| pyruvate phosphate dikinase | LinJ11_V3.1000 | 1,8 b | LmjF11.1000 | 1,3 |
| nuclear receptor binding factor-like protein | LinJ05_V3.0520 | 2,1 c | LmjF05.0520 | 1,3 |
| quinone oxidoreductase | LinJ03_V3.0550 | 11,7 c | LmjF03.0570 | 1,0 |
| quinonoid dihydropteridine reductase | LinJ34_V3.4270 | 20,2 c | LmjF34.4330 | 1,0 |
| Response to oxidative stress | ||||
| ascorbate-dependent peroxidase | LinJ34_V3.0070 | 3,0 b | LmjF34.0070 | 1,6 |
| Carbohydrate metabolic process | ||||
| 6-phosphogluconate dehydrogenase, decarboxylating | LinJ35_V3.3390 | 2,0 b | LmjF35.3340 | 1,0 |
| pyruvate kinase | LinJ35_V3.5450 | 1,9 b | LmjF35.0030 | 1,4 |
| Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | ||||
| thymidine kinase | LinJ21_V3.1450 | 1,7 b | LmjF21.1210 | 1,3 |
| DNA-directed RNA polymerase, alpha subunit | LinJ19_V3.0660 | 1,9 c | LmjF19.0660 | 1,0 |
| nuclear cap binding protein | LinJ30_V3.0560 | 4,1 c | LmjF30.0540 | 1,1 |
| vacuolar ATP synthase subunit | LinJ12_V3.0480 | 1,7 c | LmjF12.0520 | 1,0 |
| DNA metabolic process | ||||
| kinetoplast DNA-associated protein | LinJ36_V3.6180 | 1,8 b | LmjF36.5920 | 1,1 |
| RNA metabolic process | ||||
| RNA binding protein | LinJ04_V3.1190 | 2,2 b | LmjF04.1170 | 1,0 |
| RNA binding protein UBP1 | LinJ25_V3.0500 | 2,1 b | LmjF25.0490 | 1,1 |
| RNA binding protein UBP2 | LinJ25_V3.0510 | 2,1 b | LmjF25.0500 | 1,1 |
| mRNA capping methyltransferase | LinJ36_V3.0130 | 7,3 c | LmjF36.0120 | 1,1 |
| pseudouridylate synthase-like | LinJ01_V3.0280 | 1,9 c | LmjF01.0280 | 1,1 |
| RNA-binding protein | LinJ17_V3.0610 | 1,7 c | LmjF17.0550 | 1,3 |
| RNase PH-like exosome associated protein 1 | LinJ20_V3.1400 | 2,0 c | LmjF20.1360 | 1,1 |
| Amino acid and derivative metabolic process | ||||
| glycine dehydrogenase | LinJ26_V3.0040 | 8,1 c | LmjF26.0030 | 1,4 |
| Protein metabolic process | ||||
| Proteolysis | ||||
| mitochondrial processing peptidase, beta subunit | LinJ35_V3.1390 | 2,1 b | LmjF35.1380 | 1,0 |
| aminopeptidase | LinJ19_V3.0150 | 1,7 c | LmjF19.0160 | 1,2 |
| aminopeptidase | LinJ33_V3.2700 | 4,7 c | LmjF33.2570 | 1,3 |
| mitochondrial ATP-dependent zinc metallopeptidase | LinJ34_V3.1130 | 3,6 c | LmjF34.1060 | 1,1 |
| Protein folding | ||||
| chaperone protein DNAJ | LinJ18_V3.1470 | 1,8 c | LmjF18.1490 | 1,1 |
| DNAJ protein | LinJ21_V3.0550 | 4,5 c | LmjF21.0490 | 1,0 |
| Protein modification process | ||||
| protein kinase | LinJ27_V3.0100 | 4,6 c | LmjF27.0100 | 1,0 |
| Electron transport | ||||
| oxidoreductase-like protein | LinJ19_V3.1490 | 1,7 b | LmjF19.1450 | 1,0 |
| trypanothione reductase | LinJ05_V3.0350 | 1,7 b | LmjF05.0350 | 1,1 |
| Transport | ||||
| mitochondrial carrier protein | LinJ35_V3.3380 | 1,8 b | LmjF35.3330 | 1,5 |
| LinJ28_V3.2050, | ||||
| zinc transporter | LinJ28_V3.2060 | 2,5 b | LmjF28.1930 | 1,2 |
| ABC transporter | LinJ29_V3.0640 | 4,8 c | LmjF29.0620 | 1,6 |
| ABC transporter | LinJ11_V3.0040 | 10,9 c | LmjF11.0040 | 1,3 |
| amino acid permease | LinJ36_V3.0450 | 30,9 c | LmjF36.0420 | 1,4 |
| pteridine transporter | LinJ06_V3.1320 | 2,8 c | LmjF06.1260 | 1,6 |
| Cellular component organization and biogenesis | ||||
| LinJ16._V3.1550, | ||||
| kinesin | LinJ16_V3.1570 | 1,8 b | LmjF16.1460 | 1,0 |
| kinesin | LinJ23_V3.0720 | 1,9 b | LmjF23.0560 | 1,1 |
| kinesin | LinJ25_V3.2050 | 3,8 c | LmjF25.1970 | 1,1 |
| Cell cycle | ||||
| cyclin-dependent kinase regulatory subunit | LinJ32_V3.3940 | 1,7 c | LmjF32.3790 | 1,0 |
| Cell communication | ||||
| phosphoinositide-binding protein | LinJ35_V3.2470 | 1,9 b | LmjF35.2420 | 1,2 |
| rab11B GTPase | LinJ32_V3.1930 | 1,8 b | LmjF32.1840 | 1,1 |
| Unclassified | ||||
| hypothetical | LinJ02_V3.0430 | 4,3 b | LmjF02.0460 | 1,4 |
| hypothetical | LinJ19_V3.0570 | 1,8 b | LmjF19.0540 | 1,3 |
| hypothetical | LinJ19_V3.0610 | 1,8 b | LmjF19.0610 | 1,1 |
| hypothetical | LinJ19_V3.1150 | 1,9 b | LmjF19.1160 | 1,2 |
| hypothetical | LinJ20_V3.0030 | 3,6 b | LmjF20.0030 | 1,1 |
| hypothetical | LinJ23_V3.0010 | 2,1 b | LmjF23.0010 | 1,6 |
| hypothetical | LinJ23_V3.1170 | 2,2 b | LmjF23.1000 | 1,3 |
| hypothetical | LinJ24_V3.2420 | 1,8 b | LmjF24.2330 | 1,2 |
| hypothetical | LinJ27_V3.1110 | 2,5 b | LmjF27.1230 | 1,3 |
| hypothetical | LinJ28_V3.0100 | 1,8 b | LmjF28.0100 | 1,3 |
| hypothetical | LinJ32_V3.0530 | 1,7 b | LmjF32.0500 | 1,3 |
| hypothetical | LinJ33_V3.1200 | 2,6 b | LmjF33.1140 | 1,1 |
| hypothetical | LinJ33_V3.2570 | 1,8 b | LmjF33.2440 | 1,0 |
| hypothetical | LinJ35_V3.0180 | 1,8 b | LmjF35.0180 | 1,3 |
| hypothetical | LinJ35_V3.4360 | 1,7 b | LmjF35.4290 | 1,2 |
| hypothetical | LinJ36_V3.5520 | 2,9 b | LmjF36.5290 | 1,1 |
| A-1 | LinJ29_V3.1020 | 2,4 c | LmjF29.0935 | 1,6 |
| dehydrogenase/oxidoreductase-like protein | LinJ02_V3.0700 | 2,0 c | LmjF02.0730 | 1,2 |
| EF hand-like protein | LinJ13_V3.1490 | 1,9 c | LmjF13.1450 | 1,3 |
| ferric reductase transmembrane protein | LinJ30_V3.2050 | 2,0 c | LmjF30.2050 | 1,2 |
| GTPase activator protein | LinJ29_V3.1670 | 7,1 c | LmjF29.1560 | 1,1 |
| leucine rich repeat protein | LinJ28_V3.2790 | 5,5 c | LmjF28.2580 | 1,0 |
| lipin | LinJ06_V3.0860 | 1,8 c | LmjF06.0830 | 1,0 |
| membrane-bound acid phosphatase | LinJ28_V3.2850 | 2,2 c | LmjF28.2650 | 1,4 |
| zinc-finger protein | LinJ30_V3.2350 | 2,4 c | LmjF30.2340 | 1,1 |
| metallopeptidase | LinJ16_V3.0850 | 8,9 c | LmjF16.0850 | 1,3 |
| hypothetical | LinJ01_V3.0820 | 2,3 c | LmjF01.0800 | 1,0 |
| hypothetical | LinJ03_V3.0060 | 3,0 c | LmjF03.0070 | 1,3 |
| hypothetical | LinJ04_V3.1150 | 7,7 c | LmjF04.1140 | 1,3 |
| hypothetical | LinJ07_V3.0390 | 2,1 c | LmjF07.0230 | 1,2 |
| hypothetical | LinJ08_V3.0170 | 1,9 c | LmjF08.0160 | 1,1 |
| hypothetical | LinJ12_V3.0710 | 1,8 c | LmjF12.1110 | 1,3 |
| hypothetical | LinJ13_V3.0230 | 2,7 c | LmjF13.0230 | 1,2 |
| hypothetical | LinJ14_V3.0560 | 24,6 c | LmjF14.0550 | 1,4 |
| hypothetical | LinJ15_V3.0510 | 8,5 c | LmjF15.0490 | 1,0 |
| hypothetical | LinJ15_V3.1000 | 4,7 c | LmjF15.0940 | 1,0 |
| hypothetical | LinJ16_V3.0620 | 5,2 c | LmjF16.0620 | 1,3 |
| hypothetical | LinJ16_V3.1260 | 11,6 c | LmjF16.1210 | 1,1 |
| hypothetical | LinJ17_V3.0600 | 2,4 c | LmjF17.0540 | 1,1 |
| hypothetical | LinJ18_V3.0120 | 1,9 c | LmjF18.0120 | 1,1 |
| hypothetical | LinJ18_V3.0300 | 3,0 c | LmjF18.0300 | 1,1 |
| hypothetical | LinJ19_V3.1170 | 9,0 c | LmjF19.1180 | 1,4 |
| hypothetical | LinJ20_V3.0450 | 1,8 c | LmjF20.0380 | 1,1 |
| hypothetical | LinJ20_V3.1670 | 3,3 c | LmjF20.1700 | 1,4 |
| hypothetical | LinJ21_V3.0920 | 4,6 c | LmjF21.0825 | 1,1 |
| hypothetical | LinJ21_V3.1220 | 5,1 c | LmjF21.0980 | 1,3 |
| hypothetical | LinJ22_V3.0110 | 1,8 c | LmjF22.0240 | 1,2 |
| hypothetical | LinJ23_V3.1520 | 2,3 c | LmjF23.1267 | 1,1 |
| hypothetical | LinJ24_V3.0700 | 8,2 c | LmjF24.0690 | 1,5 |
| hypothetical | LinJ24_V3.1080 | 4,0 c | LmjF24.1060 | 1,1 |
| hypothetical | LinJ24_V3.1620 | 1,9 c | LmjF24.1550 | 1,1 |
| hypothetical | LinJ25_V3.0220 | 11,0 c | LmjF25.0220 | 1,2 |
| hypothetical | LinJ25_V3.0460 | 5,0 c | LmjF25.0450 | 1,4 |
| hypothetical | LinJ25_V3.0560 | 32,7 c | LmjF25.0550 | 1,1 |
| hypothetical | LinJ26_V3.1260 | 1,9 c | LmjF26.1280 | 1,0 |
| hypothetical | LinJ26_V3.1850 | 2,8 c | LmjF26.1850 | 1,1 |
| hypothetical | LinJ26_V3.2220 | 6,5 c | LmjF26.2210 | 1,2 |
| hypothetical | LinJ27_V3.1080 | 2,8 c | LmjF27.1200 | 1,1 |
| hypothetical | LinJ29_V3.0930 | 46,7 c | LmjF29.0868 | 1,5 |
| hypothetical | LinJ30_V3.0160 | 12,4 c | LmjF30.0170 | 1,2 |
| hypothetical | LinJ31_V3.0490 | 5,4 c | LmjF31.0470 | 1,3 |
| hypothetical | LinJ31_V3.1630 | 2,2 c | LmjF31.1600 | 1,0 |
| hypothetical | LinJ32_V3.1410 | 2,2 c | LmjF32.1350 | 1,1 |
| hypothetical | LinJ32_V3.3760 | 2,9 c | LmjF32.3600 | 1,2 |
| hypothetical | LinJ33_V3.0590 | 1,8 c | LmjF33.0565 | 1,3 |
| hypothetical | LinJ33_V3.0810 | 2,1 c | LmjF33.0760 | 1,1 |
| hypothetical | LinJ33_V3.2850 | 1,9 c | LmjF33.2710 | 1,4 |
| hypothetical | LinJ34_V3.2250 | 2,0 c | LmjF34.2473 | 1,2 |
| hypothetical | LinJ34_V3.3520 | 4,1 c | LmjF34.3730 | 1,1 |
| hypothetical | LinJ35_V3.4090 | 10,3 c | LmjF35.4040 | 1,2 |
| hypothetical | LinJ35_V3.1640 | 9,4 c | LmjF35.1640 | 1,2 |
| hypothetical | LinJ35_V3.5350 | 2,1 c | LmjF35.5030 | 1,0 |
| hypothetical | LinJ36_V3.0470 | 6,3 c | LmjF36.0440 | 1,1 |
| hypothetical | LinJ36_V3.4410 | 3,5 c | LmjF36.4200 | 1,6 |
| hypothetical | LinJ36_V3.4940 | 1,8 c | LmjF36.4710 | 1,1 |
a Only non-modulated genes are listed in this column.
b Genes differentially expressed in promastigotes.
c Genes differentially expressed in amastigotes.
Table 4.
Genes differentially expressed in Leishmania major but constitutively expressed in Leishmania infantum.
| GO annotation (molecular function) | Accession number (GeneDB) | Fold increase a | Accession number (GeneDB) | Fold increase |
| L. infantum | L. major | |||
| Metabolic process | ||||
| 2-aminoethylphosphonate: pyruvateaminotransferase | LinJ03_V3.0400 | 1,0 | LmjF03.0040 | 2,0 b |
| folylpolyglutamate synthetase | LinJ36_V3.2740 | 1,1 | LmjF36.2610 | 2,1 b |
| phosphatidylethanolaminen-methyltransferase | LinJ31_V3.3250 | 1,3 | LmjF31.3120 | 2,4 b |
| aldehyde dehydrogenase | LinJ25_V3.1160 | 1,1 | LmjF25.1120 | 3,7 c |
| arginase | LinJ35_V3.1490 | 1,3 | LmjF35.1480 | 2,8 c |
| Carbohydrate metabolic process | ||||
| 6-phospho-1-fructokinase | LinJ29_V3.2620 | 1,4 | LmjF29.2510 | 2,6 b |
| aldose 1-epimerase | LinJ35_V3.0990 | 1,1 | LmjF35.0970 | 3,2 b |
| beta-fructofuranosidase | LinJ04_V3.0300 | 1,3 | LmjF04.0310 | 5,4 b |
| beta-fructofuranosidase | LinJ04_V3.0310 | 1,2 | LmjF04.0320 | 3,6 b |
| LinJ35_V3.0640, | ||||
| beta-fructofuranosidase | LinJ35_V3.0650 | 1,6 | LmjF35.0640 | 5,9 b |
| LinJ21_V3.0300, | LmjF21.0240, | |||
| hexokinase | LinJ21_V3.0310 | 1,2 | LmjF21.0250 | 2,2 b |
| glyceraldehyde 3-phosphate dehydrogenase | LinJ36_V3.2480 | 1,4 | LmjF36.2350 | 3,6 c |
| Lipid metabolic process | ||||
| farnesyl pyrophosphate synthase | LinJ22_V3.1210 | 1,2 | LmjF22.1360 | 1,9 b |
| fatty acid elongase | LinJ14_V3.0670 | 1,6 | LmjF14.0650 | 2,7 b |
| mevalonate kinase | LinJ31_V3.0580 | 1,1 | LmjF31.0560 | 2,1 b |
| phospholipase c-like | LinJ30_V3.2970 | 1,3 | LmjF30.2950 | 2,1 c |
| polyprenyl synthase | LinJ19_V3.0210 | 1,0 | LmjF19.0220 | 2,4 c |
| Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | ||||
| adenosine deaminase | LinJ35_V3.2200 | 1,1 | LmjF35.2160 | 4,6 b |
| DNA metabolic process | ||||
| j-binding protein | LinJ14_V3.0040 | 1,4 | LmjF14.0040 | 2,3 b |
| RNA metabolic process | ||||
| RNA-binding protein | LinJ23_V3.0900 | 1,3 | LmjF23.0730 | 3,6 c |
| Amino acid and derivative metabolic process | ||||
| tyrosine aminotransferase | LinJ36_V3.2490 | 1,1 | LmjF36.2360 | 2,7 b |
| Protein metabolic process | ||||
| lipophosphoglycan biosynthetic protein | LinJ29_V3.0790 | 1,3 | LmjF29.0760 | 2,3 b |
| Proteolysis | ||||
| calpain-like cysteine peptidase | LinJ20_V3.1230 | 1,1 | LmjF20.1190 | 2,6 b |
| calpain-like cysteine peptidase | LinJ27_V3.0510 | 1,3 | LmjF27.0510 | 2,8 b |
| carboxypeptidase | LinJ33_V3.2670 | 1,6 | LmjF33.2540 | 3,0 b |
| carboxypeptidase | LinJ13_V3.0090 | 1,1 | LmjF13.0090 | 2,1 b |
| glutamamyl carboxypeptidase | LinJ29_V3.1680 | 1,1 | LmjF29.1570 | 1,7 b |
| oligopeptidase b | LinJ09_V3.0820 | 1,1 | LmjF09.0770 | 2,2 b |
| pyroglutamyl-peptidase (PGP) | LinJ34_V3.1750 | 1,4 | LmjF34.2000 | 2,0 b |
| cysteine protease | LinJ19_V3.1460 | 1,3 | LmjF19.1420 | 3,1 c |
| Protein folding | ||||
| chaperone protein DNAJ | LinJ32_V3.3220 | 1,1 | LmjF32.3030 | 2,2 b |
| Protein modification process | ||||
| protein kinase | LinJ26_V3.2600 | 1,2 | LmjF26.2570 | 4,3 b |
| protein kinase | LinJ21_V3.0190 | 1,1 | LmjF21.0130 | 2,2 b |
| mitogen-activated protein kinase 3 | LinJ10_V3.0540 | 1,0 | LmjF10.0490 | 2,0 b |
| serine/threonine-protein kinase | LinJ31_V3.3070 | 1,1 | LmjF31.2960 | 2,8 b |
| serine/threonine-protein phosphatase PP1 | LinJ34_V3.0850 | 1,1 | LmjF34.0810 | 1,8 b |
| protein kinase | LinJ36_V3.4460 | 1,6 | LmjF36.4250 | 1,7 c |
| Electron transport | ||||
| D-lactate dehydrogenase | LinJ29_V3.0290 | 1,1 | LmjF29.0280 | 3,0 b |
| pyrroline-5-carboxylate reductase | LinJ13_V3.1420 | 1,6 | LmjF13.1680 | 2,1 b |
| UDP-galactopyranose | LinJ18_V3.0200 | 1,1 | LmjF18.0200 | 1,7 b |
| Transport | ||||
| amino acid permease | LinJ27_V3.0530 | 1,6 | LmjF27.0680 | 3,0 b |
| biopterin transporter | LinJ35_V3.5120 | 1,1 | LmjF35.5150 | 2,4 b |
| LmjF36.6280, | ||||
| glucose transporter, lmgt2 | LinJ36_V3.6550 | 1,0 | LmjF36.6290 | 5,6 b |
| pteridine transporter | LinJ06_V3.0310 | 1,1 | LmjF06.0310 | 3,2 b |
| sugar transporter | LinJ24_V3.0690 | 1,0 | LmjF24.0680 | 3,9 b |
| transmembrane amino acid transporter | LinJ07_V3.1340 | 1,1 | LmjF07.1160 | 9,7 b |
| LinJ15_V3.1230, | ||||
| LinJ15_V3.1240, | ||||
| LinJ15_V3.1250, | LmjF15.1230, | |||
| nucleoside transporter 1 | LinJ15_V3.1260 | 1,2 | LmjF15.1240 | 4,5 b |
| mitochondrial ornithine transporter 1-like | LinJ16_V3.0220 | 1,0 | LmjF16.0210 | 2,2 b |
| Cell component organization and biogenesis | ||||
| dynein heavy chain | LinJ36_V3.1010 | 1,6 | LmjF36.0950 | 2,6 b |
| kinesin | LinJ14_V3.0870 | 1,1 | LmjF14.0810 | 1,8 b |
| kinesin | LinJ21_V3.1280 | 1,4 | LmjF21.1040 | 1,9 b |
| LmjF09.0150, | ||||
| LmjF09.0154, | ||||
| LmjF09.0158, | ||||
| LmjF09.0162, | ||||
| LmjF09.0166, | ||||
| LmjF09.0170, | ||||
| microtubule associated protein | LinJ09_V3.0180 | 1,3 | LmjF09.0174 | 2,5 b |
| LinJ19_V3.0820, | ||||
| microtubule associated protein | LinJ19_V3.0850 | 1,1 | LmjF19.0860 | 2,5 b |
| PFR | LinJ05_V3.0920 | 1,3 | LmjF05.0920 | 2,3 b |
| PFR par4 | LinJ05_V3.0040 | 1,3 | LmjF05.0040 | 2,8 b |
| Cell communication | ||||
| cAMP specific phosphodiesterase | LinJ15_V3.1550 | 1,5 | LmjF15.1480 | 1,8 b |
| phosphodiesterase | LinJ18_V3.1100 | 1,4 | LmjF18.1090 | 2,3 b |
| Cell motility | ||||
| myosin heavy chain | LinJ32_V3.4020 | 1,6 | LmjF32.3870 | 3,5 b |
| Unclassified | ||||
| acid phophatase | LinJ36_V3.2600 | 1,0 | LmjF36.2470 | 1,9 b |
| long chain fatty Acyl CoA synthetase | LinJ03_V3.0220 | 1,1 | LmjF03.0230 | 1,9 b |
| LinJ19_V3.1350, | LmjF19.1340, | |||
| glycerol uptake protein | LinJ19_V3.1360 | 1,3 | LmjF19.1345 | 1,8 b |
| membrane-bound acid phosphatase 2 | LinJ36_V3.2720 | 1,1 | LmjF36.2590 | 4,6 b |
| nons pecific nucleoside hydrolas e | LinJ18_V3.1570 | 1,1 | LmjF18.1580 | 2,1 b |
| prostaglandin f2-alpha synthase | LinJ31_V3.2210 | 1,5 | LmjF31.2150 | 5,1 b |
| LinJ31_V3.0950, | Lm jF 31.0920, | |||
| s odium s tibogluconate res is tance protein | LinJ31_V3.3400 | 1,4 | LmjF31.0950 | 2,1 b |
| s urfac e antigen-lik e | LinJ04_V3.0170 | 1,0 | Lm jF04.0180 | 2,0 b |
| s urfac e antigen-lik e | LinJ04_V3.0180 | 1,0 | Lm jF04.0190 | 2,7 b |
| Lm jF 12.0850, | ||||
| Lm jF 12.0860, | ||||
| Lm jF 12.0870, | ||||
| surface antigen 2 | LinJ12_V3.0020 | 1,2 | LmjF12.0890 | 2,1 b |
| ubiquitin-conjugating enzy me-like | LinJ21_V3.0500 | 1,6 | Lm jF21.0440 | 2,2 b |
| hypothetical | LinJ03_V3.0340 | 1,4 | Lm jF03.0360 | 1,9 b |
| Lm jF 04.0130, | ||||
| Lm jF 04.0140, | ||||
| Lm jF 04.0150, | ||||
| Lm jF 04.0160, | ||||
| hypothetical | LinJ04_V3.0160 | 1,1 | LmjF04.0170 | 2,9 b |
| hypothetical | LinJ05_V3.1070 | 1,3 | Lm jF05.1070 | 3,0 b |
| hypothetical | LinJ09_V3.1360 | 1,0 | Lm jF09.1300 | 2,0 b |
| hypothetical | LinJ09_V3.1600 | 1,4 | Lm jF09.1510 | 2,9 b |
| hypothetical | LinJ09_V3.1610 | 1,1 | Lm jF09.1520 | 3,2 b |
| Lm jF 11.0670, | ||||
| LinJ11_V3.0680, | Lm jF 11.0673, | |||
| hypothetical | LinJ11_V3.0690 | 1,0 | LmjF11.0675 | 3,0 b |
| hypothetical | LinJ04_V3.1220 | 1,3 | Lm jF04.1200 | 2,0 b |
| LinJ14_V3.0490, | Lm jF 14.0480, | |||
| hypothetical | LinJ14_V3.0500 | 1,3 | LmjF14.0490 | 2,6 b |
| hypothetical | LinJ15_V3.0560 | 1,0 | Lm jF15.0540 | 1,9 b |
| hypothetical | LinJ17_V3.0690 | 1,4 | Lm jF17.0630 | 2,5 b |
| hypothetical | LinJ17_V3.0990 | 1,1 | Lm jF17.0890 | 6,5 b |
| hypothetical | LinJ18_V3.1300 | 1,1 | Lm jF18.1320 | 1,9 b |
| hypothetical | LinJ19_V3.0070 | 1,4 | Lm jF19.0080 | 3,5 b |
| hypothetical | LinJ23_V3.0890 | 1,3 | Lm jF23.0720 | 2,4 b |
| hypothetical | LinJ23_V3.1020 | 1,1 | Lm jF23.0840 | 2,6 b |
| hypothetical | LinJ23_V3.1730 | 1,4 | Lm jF23.1690 | 1,8 b |
| hypothetical | LinJ24_V3.1110 | 1,1 | Lm jF24.1090 | 4,6 b |
| hypothetical | LinJ25_V3.2090 | 1,1 | Lm jF25.2010 | 2,7 b |
| hypothetical | LinJ26_V3.1980 | 1,5 | Lm jF26.1980 | 1,9 b |
| hypothetical | LinJ28_V3.0220 | 1,1 | LmjF28.0220 | 1,7 b |
| hypothetical | LinJ28_V3.0420 | 1,2 | LmjF28.0280 | 1,8 b |
| hypothetical | LinJ29_V3.1820 | 1,0 | Lm jF29.1690 | 2,8 b |
| hypothetical | LinJ29_V3.2550 | 1,6 | Lm jF29.2440 | 3,3 b |
| hypothetical | LinJ30_V3.1230 | 1,1 | Lm jF30.1170 | 2,5 b |
| hypothetical | LinJ30_V3.2700 | 1,3 | Lm jF30.2700 | 3,3 b |
| hypothetical | LinJ32_V3.0480 | 1,3 | LmjF32.0470 | 2,0 b |
| hypothetical | LinJ32_V3.1760 | 1,4 | Lm jF32.1680 | 3,3 b |
| hypothetical | LinJ32_V3.3350 | 1,3 | Lm jF32.3150 | 2,3 b |
| hypothetical | LinJ32_V3.3650 | 1,3 | Lm jF32.3450 | 1,9 b |
| hypothetical | LinJ33_V3.0220 | 1,6 | Lm jF33.0210 | 4,0 b |
| hypothetical | LinJ33_V3.1040 | 1,0 | Lm jF33.0990 | 3,4 b |
| hypothetical | LinJ33_V3.1130 | 1,4 | LmjF33.1070 | 2,7 b |
| hypothetical | LinJ33_V3.2940 | 1,4 | LmjF33.2800 | 1,7 b |
| hypothetical | LinJ34_V3.0210 | 1,1 | LmjF34.0190 | 2,1 b |
| hypothetical | LinJ34_V3.0740 | 1,2 | LmjF34.0705 | 1,8 b |
| hypothetical | LinJ34_V3.4110 | 1,0 | LmjF34.4280 | 2,4 b |
| hypothetical | LinJ35_V3.1470 | 1,0 | LmjF35.1460 | 1,7 b |
| hypothetical | LinJ35_V3.5400 | 1,2 | LmjF35.5080 | 1,7 b |
| hypothetical | LinJ36_V3.0730 | 1,0 | LmjF36.0670 | 2,5 b |
| hypothetical | LinJ36_V3.1190 | 1,0 | LmjF36.1130 | 1,7 b |
| hypothetical | LinJ36_V3.1230 | 1,3 | LmjF36.1170 | 2,1 b |
| hypothetical | LinJ36_V3.1520 | 1,1 | LmjF36.1460 | 1,7 b |
| hypothetical | LinJ36_V3.4470 | 1,1 | LmjF36.4260 | 2,0 b |
| Lm jF30.1410, | ||||
| Lm jF30.1420, | ||||
| ama 1 | LinJ30_V3.1490 | 1,1 | LmjF30.1430 | 2,1 c |
| Lm jF31.1450, | ||||
| s urfac e membrane protein gp46-like | LinJ31_V3.1490 | 1,1 | LmjF31.1460 | 2,1 c |
| hypothetical | LinJ06_V3.0030 | 1,1 | LmjF06.0030 | 2,0 c |
| hypothetical | LinJ17_V3.0390 | 1,3 | LmjF17.0340 | 4,0 c |
| hypothetical | LinJ18_V3.1110 | 1,1 | LmjF18.1100 | 1,7 c |
| hypothetical | LinJ19_V3.1540 | 1,1 | LmjF19.1490 | 1,8 c |
| hypothetical | LinJ20_V3.1630 | 1,1 | LmjF20.1660 | 2,0 c |
| hypothetical | LinJ24_V3.0590 | 1,2 | LmjF24.0580 | 4,8 c |
| hypothetical | LinJ25_V3.0230 | 1,3 | LmjF25.0230 | 2,0 c |
| hypothetical | LinJ26_V3.1500 | 1,3 | LmjF26.1520 | 3,5 c |
| hypothetical | LinJ28_V3.1070 | 1,4 | LmjF28.0980 | 2,6 c |
| hypothetical | LinJ28_V3.2860 | 1,2 | LmjF28.2660 | 2,0 c |
| hypothetical | LinJ31_V3.2910 | 1,4 | LmjF31.2810 | 2,1 c |
| hypothetical | LinJ32_V3.2620 | 1,3 | LmjF32.2480 | 1,8 c |
| hypothetical | LinJ35_V3.4870 | 1,3 | LmjF35.4810 | 1,8 c |
| hypothetical | LinJ36_V3.2960 | 1,1 | LmjF36.2820 | 1,9 c |
| hypothetical | LinJ36_V3.3480 | 1,3 | LmjF36.3320 | 1,7 c |
| hypothetical | LinJ36_V3.4870 | 1,0 | LmjF36.4640 | 2,0 c |
a Only non-modulated genes are listed in this column.
b Genes differentially expressed in promastigotes.
c Genes differentially expressed in amastigotes.
Confirmation of gene expression patterns by quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was used to validate the microarray results. Changes in expression levels of 80 selected L. infantum and/or L. major mRNAs in either promastigotes or intracellular amastigotes as determined by the microarray experiments were confirmed by qRT-PCR. Genes for qRT-PCR were randomly selected based on different criteria, which included high vs. lower levels of expression, constitutive vs. stage-regulated gene expression, different GO categories or genes encoding unclassified proteins, single copy genes and genes belonging to larger gene families. mRNA expression levels determined by qRT-PCR were normalized to three L. infantum and L. major constitutively expressed mRNAs (see Methods). Estimated expression patterns by qRT-PCR were compared to those by DNA microarrays. Results obtained by qRT-PCR were consistent with the microarray data in more than 97% of the cases (Table 5). In few cases (~10%), the difference in expression levels between the two methods (e.g. DNA microarray vs. qRT-PCR) was two-fold or higher, however, both methods agreed on the differential gene expression in the same life cycle stage (Table 5).
Table 5.
Comparison of microarray mRNA expression levels to relative expression levels determined by quantitative real time PCR (qRT-PCR).
| Accession number (GeneDB) | Ratio Ama/Pro | |
| DNA Microarrays a | qRT-PCR b | |
| L. infantum | ||
| LinJ30_V 3.2870 | 0,2 | 0,5 ± 0,0 |
| LinJ34_V 3.0070 | 0,3 | 0,5 ± 0,0 |
| LinJ36_V 3.5620 | 0,4 | 0,3 ± 0,0 |
| LinJ21_V 3.1490 | 0,4 | 0,5 ± 0,1 |
| LinJ14_V 3.1240 | 0,4 | 0,5 ± 0,1 |
| LinJ23_V 3.0860 | 0,5 | 0,3 ± 0,0 |
| LinJ25_V 3.0500 | 0,5 | 0,2 ± 0,2 |
| LinJ16_V 3.0950 | 0,5 | 0,5 ± 0,0 |
| LinJ18_V 3.1090 | 0,5 | 0,6 ± 0,0 |
| LinJ24_V 3.0870 | 0,6 | 0,5 ± 0,0 |
| LinJ30_V 3.1520 | 1,7 | 2,2 ± 0,3 |
| LinJ33_V 3.2470 | 1,8 | 1,3 ± 0,1 |
| LinJ34_V 3.1730 | 2,0 | 2,3 ± 0,3 |
| LinJ30_V 3.2050 | 2,0 | 1,8 ± 0,1 |
| LinJ34_V 3.1160 | 2,3 | 1,9 ± 0,6 |
| LinJ31_V 3.0370 | 2,4 | 2,2 ± 0,3 |
| LinJ14_V 3.1450 | 2,4 | 3,7 ± 0,3 |
| LinJ34_V 3.1020 c | 2,8 | 10,1 ± 0,5 |
| LinJ30_V 3.0560 | 4,1 | 3,3 ± 0,3 |
| LinJ27_V 3.0100 | 4,6 | 3,7 ± 0,4 |
| LinJ19_V 3.0420 | 5,0 | 3,0 ± 0,3 |
| LinJ36_V 3.6530 | 5,0 | 1,9 ± 0,2 |
| LinJ23_V 3.1790 | 7,0 | 2,4 ± 0,5 |
| LinJ36_V 3.0130 | 7,3 | 4,2 ± 0,3 |
| LinJ31_V 3.2790 | 7,6 | 2,0 ± 0,1 |
| LinJ26_V 3.0040 | 8,1 | 2,4 ± 0,2 |
| LinJ36_V 3.4180 | 8,7 | 6,1 ± 0,6 |
| LinJ34_V 3.4270 | 20,2 | 2,4 ± 0,2 |
| LinJ14_V 3.1500 | 22,0 | 1,2 ± 0,1 |
| LinJ14_V 3.0560 | 24,6 | 2,0 ± 0,0 |
| LinJ36_V 3.2480 | 0,7 | 0,6 ± 0,3 |
| LinJ14_V 3.0760 | 0,9 | 1,5 ± 0,1 |
| LinJ25_V 3.1160 | 0,9 | 1,0 ± 0,1 |
| LinJ07_V 3.0550 | 1,0 | 1,6 ± 0,2 |
| LinJ28_V 3.0470 | 1,0 | 1,2 ± 0,1 |
| LinJ34_V 3.1910 | 1,0 | 1,0 ± 0,5 |
| LinJ19_V 3.0010 | 1,2 | 1,6 ± 0,1 |
| LinJ34_V 3.2420 | 1,2 | 1,3 ± 0,1 |
| LinJ30_V 3.2200 | 1,3 | 1,3 ± 0,1 |
| LinJ36_V 3.4000 | 1,4 | 1,8 ± 0,3 |
| LinJ17_V 3.0440 | 1,5 | 3,0 ± 0,2 |
| LinJ08_V 3.1220 | 1,6 | 1,7 ± 0,1 |
| L. major | ||
| Lm jF33.2340 | 0,5 | 0,3 ± 0,0 |
| Lm jF36.1360 | 0,5 | 0,8 ± 0,0 |
| Lm jF29.2510 | 0,4 | 0,02 ± 0,00 |
| Lm jF36.2360 | 0,4 | 0,4 ± 0,0 |
| Lm jF06.0310 | 0,3 | 0,4 ± 0,0 |
| Lm jF35.0970 | 0,3 | 0,6 ± 0,0 |
| Lm jF23.0690 | 0,3 | 0,2 ± 0,0 |
| Lm jF23.1300 | 0,3 | 0,1 ± 0,0 |
| Lm jF02.0160 | 0,3 | 0,1 ± 0,0 |
| Lm jF23.0710 | 0,2 | 0,1 ± 0,0 |
| Lm jF35.2160 | 0,2 | 0,1 ± 0,0 |
| Lm jF36.2590 | 0,2 | 0,1 ± 0,0 |
| Lm jF21.0240 | 0,2 | 0,2 ± 0,0 |
| Lm jF31.0350 | 0,2 | 0,1 ± 0,0 |
| Lm jF04.0310 | 0,2 | 0,3 ± 0,0 |
| Lm jF07.1160 | 0,1 | 0,1 ± 0,0 |
| Lm jF31.2460 | 2,6 | 2,3 ± 0,2 |
| Lm jF28.2910 | 2,8 | 1,9 ± 0,2 |
| Lm jF36.5960 | 3,1 | 2,0 ± 2,1 |
| Lm jF23.0730 | 3,6 | 1,1 ± 0,1 |
| Lm jF36.2350 | 3,6 | 4,5 ± 0,4 |
| Lm jF25.1120 | 3,7 | 2,3 ± 0,3 |
| Lm jF08.0820 | 4,3 | 6,6 ± 0,9 |
| Lm jF14.1360 | 4,7 | 4,4 ± 0,4 |
| Lm jF30.2190 | 5,8 | 3,0 ± 0,1 |
| Lm jF11.1220 | 6,7 | 5,2 ± 0,3 |
| Lm jF34.1840 | 7,3 | 3,1 ± 0,2 |
| Lm jF35.4230 | 10,3 | 1,9 ± 0,1 |
| Lm jF31.3190 | 10,9 | 3,7 ± 0,4 |
| Lm jF36.3810 | 1,0 | 1,1 ± 0,1 |
| Lm jF28.0330 | 1,1 | 1,3 ± 0,2 |
| Lm jF30.2050 | 1,2 | 1,5 ± 0,2 |
| Lm jF03.0570 | 1,0 | 1,2 ± 0,2 |
| Lm jF34.4510 | 1,0 | 2,0 ± 0,2 |
| Lm jF34.0070 | 1,6 | 1,0 ± 0,1 |
a The microarray results have all a p-value lower than 0.05.
b The same RNA preparations were used for the microarray and qRT-PCR experiments. Levels of mRNA determined by qRT-PCR were normalized as described in Methods. The values reported here are the average of two biological replicates and three technical replicates. The set of primers for qRT-PCR amplification are presented in Additional file 6.
c LinJ34_V3.1020 encodes an amastin homologue that shares homology to other members of the amastin gene family. For the microarray experiment, the 70-mer probe used recognizes more than one amastin members whereas the set of primers used for qRT-PCR was specific to the LinJ34_V3.1020 gene only.
d LinJ14_V3.1500 is a member of the glycosylation phosphoglycan beta 1,3 galactosyltransferase gene family. For the microarray experiment the 70-mer probe used recognizes more than one gene family members whereas the set of primers used for qRT-PCR was specific to the LinJ14_V3.1500 gene only, whose expression was not modulated.
In this study, L. infantum amastigotes were isolated from THP1-infected macrophages and L. major amastigotes from mouse lesions. Our attempts to isolate sufficient amastigote RNA material from the spleen/liver of infected hamsters were unsuccessful due to low infection rates obtained with the L. infantum strain we used in this study. To investigate whether gene expression levels could be different in lesion- vs. THP1-derived amastigotes, we first evaluated by qRT-PCR analysis the expression patterns of a selected number of transcripts between L. major amastigotes isolated from mouse lesions or THP1 infected cells in vitro. The majority of the genes tested (73%) were modulated in a similar fashion in THP1-derived or lesion-derived L. major amastigotes (see Additional file 5). Even when L. major or L. infantum amastigotes were grown within THP1-infected cells, different expression patterns were obtained for the majority (67%) of the transcripts analyzed by qRT-PCR (see Additional file 5). These results further support our microarray data demonstrating substantial differences in amastigote-regulated gene expression between L. major and L. infantum (Figure 3).
Stage-regulated transcripts are distributed throughout the Leishmania spp. chromosomes
As mentioned in the Introduction, up to several hundred Leishmania genes are co-transcribed into polycistronic RNAs, and individual mature mRNAs are resolved by trans-splicing and polyadenylation [22,23]. To determine genome distribution and chromosomal organization of the differentially expressed genes in both Leishmania species, global RNA expression profiles for both developmental stages of L. major and L. infantum were generated for each of the 36 chromosomes (GeneDB, [19,20]). As representative examples, only chromosomes 5, 16, 30 and 36 are shown here (Figure 4). This global genome analysis revealed that differentially expressed genes in either life stage are randomly distributed throughout Leishmania chromosomes and that there is apparently no clustering of these genes within specific genomic loci (Figure 4 and data not shown). These observations are consistent with published work to date and further support the concept that stage-regulated gene expression in Leishmania involves mostly post-transcriptional mechanisms.
Figure 4.

Distribution of differentially expressed transcripts in chromosomes 5, 16, 30 and 36 of Leishmania major and Leishmania infantum. On the horizontal axis are all genes in the order found on the selected chromosomes according to GeneDB [24]. On the vertical axis is the amastigote (Ama) to promastigote (Pro) expression ratio. Genes upregulated in the amastigote stage are above the line at 0.75 and genes upregulated in promastigotes are below the line -0.75. Blue dots represent stage-regulated expression of genes on the respective L. infantum chromosomes and black dots represent differential expression of genes on the same chromosomes of L. major.
SIDER retroposons and their distribution in the 3'UTR of Leishmania differentially expressed transcripts
We recently identified two large classes of distinct short interspersed degenerate retroposons, named SIDER1 (~785 copies) and SIDER2 (~1073 copies) that are mainly located in the 3'UTR of Leishmania mRNAs [46]. Members of the SIDER1 subfamily correspond to the previously identified ~450 nt 3'UTR element conserved in several developmentally regulated mRNAs, including the amastin mRNAs [15,47]. SIDER1 was shown to regulate amastin mRNA translation in a stage-specific manner [47,48] whereas SIDER2 plays a rather global role in mRNA degradation [46]. Since SIDERs and especially SIDER2 are involved in the regulation of mRNA stability, we investigated whether these widespread retroposons were associated more frequently with differentially expressed mRNAs. Therefore, we screened all differentially expressed transcripts as determined by microarray experiments in both Leishmania species for the presence of SIDER1 or SIDER2 homologous sequences in their potential 3'UTR (putative 3'UTRs were mapped using bioinformatics tools as described [46,49]). On average, 21% of the promastigote-upregulated transcripts and 26–42% of the amastigote-upregulated transcripts in either species contained either SIDER1 or SIDER2 in their 3'UTR (Table 6). The higher percentage of SIDER1 in amastigotes can be partly explained by the presence of this subclass of retroposons in amastin mRNAs that are part of a large gene family ([15], unpublished data)). Interestingly, L. major amastigote-upregulated transcripts contained generally a higher percentage of SIDER1/2 retroposons and especially SIDER1 compared to L. infantum (42% vs. 26%). Considering that ~25% of the L. major or L. infantum transcripts bear either SIDER1 or SIDER2 in their 3'UTR (Table 6) ([46] (Smith, M. et al., unpublished)), our data indicate that SIDERs, at least in L. infantum, are not preferentially associated with differentially upregulated transcripts, which suggests that additional regulatory elements may also contribute to the stage-specific accumulation of Leishmania mRNAs.
Table 6.
Distribution of SIDER retroposons among differentially expressed Leishmania spp. transcripts.
| Differentially Expressed Transcriptsa | Genomic Distributionb | ||||
| Species | Life Stage | SIDER1% | SIDER2% | SIDER1% | SIDER2% |
| L. major | amastigote | 23.9 | 18.3 | 11.6 | 15.0 |
| promastigote | 10.6 | 11.6 | |||
| L. infantum | amastigote | 14.1 | 11.9 | 9.4 | 15.8 |
| promastigote | 8.4 | 12.4 | |||
a As determined by the DNA microarray experiments (this study).
b Refined species-specific hidden Markov model profiles were used to determine the genomic distribution of two SIDER families in L. infantum and L. major ([46]; Smith et al. unpublished)). Values represent the proportion of annotated coding sequences that potentially harbor at least one SIDER element in their 3'UTR. The percentage of SIDERs in differentially expressed transcripts is compared with the genomic distribution of SIDERs.
Discussion
This study provides an extensive analysis of genome-wide expression profiling of the main developmental life stages (e.g. procyclic promastigotes and intracellular amastigotes) of L. infantum along with a comparative analysis of gene expression profiles between L. infantum and L. major, two species causing distinct pathologies in humans, using a multispecies Leishmania DNA microarray. The comparative analysis between L. major and L. infantum transcriptomes is the first to date and may lead to a better understanding of how differential gene expression among species with high similarity in genome sequences may be involved in the development of different disease phenotypes.
Expression patterns, stage-regulated genes and pathways identified
Genes whose expression was upregulated in either promastigote or amastigote life stages of L. major and L. infantum, as determined by DNA microarrays, belong to various biological processes. Approximately 25% of the differentially expressed genes between both life stages in both species are involved in metabolism (Figure 2). Promastigotes and amastigotes reside in different environments and it is therefore expected that their metabolic needs will differ. Promastigotes in the sand fly use glycolysis and mitochondrial metabolism as their main energy sources (reviewed in [50]). Amastigotes contain glycosomes, although considerably fewer than promastigotes [50]. L. mexicana amastigotes isolated from mouse lesions have a reduced need for proline and glucose consumption and increased beta-oxidation of fatty acids compared to promastigotes, which could be used as energy source [51,52]. Our study revealed indeed that genes involved in carbohydrate metabolism as well as several glucose transporters were overexpressed in promastigotes compared to amastigotes (Table 2, see Additional files 1 and 1). Interestingly, recent proteomic studies using axenic L. donovani promastigote and amastigote cultures indicated that the differentiating parasite shifts from glucose to fatty acids and amino acids as its main energy source [53]. Enzymes required for the de novo synthesis of inositol and mannose are important for amastigote growth [54]. In agreement with this, the myo-inositol-1-phosphate synthetase gene encoding a key enzyme in the first step of inositol synthesis was found upregulated in both L. major and L. infantum amastigotes (Table 2).
The absence of the glyoxylate pathway in Leishmania, which is required for the conversion of acetyl-CoA into sugars, indicates that amastigotes may be unable to utilize fatty acids as their major carbon source [19,55]. Amino acids are the second carbon source for Leishmania promastigotes but are also important in the amastigote stage [56-58]. The phagolysosomes of macrophages contain high levels of amino acids generated by proteolytic digestion of host phagosomal proteins and from exogenous proteins delivered into the phagolysosome via endocytosis [54,59]. Consistently, several lysosomal aminopeptidases and cathepsin-L like cysteine proteinases were upregulated in amastigotes (see Additional files 2 and 4). Also, several amino acid transporters and permeases important for the transport of amino acids from the phagosome into amastigotes were found upregulated in amastigotes (see Additional files 2 and 4 and [58]). Leishmania amastigotes can scavenge all their purine requirements, cations, vitamins, carbon sources and several essential amino acids from the macrophage phagolysosome via plasma membrane transporters [56]. Consistent with this, intracellular amastigotes of both L. major and L. infantum overexpress a relatively larger number of membrane transporters than promastigotes (see Additional files 2 and 4).
Important differences were observed in the expression of genes involved in cellular organization, biogenesis and cell motility between promastigote and amastigote stages of both Leishmania species. The motile flagellated promastigotes as opposed to the aflagellated amastigotes overexpress dyneins corresponding to large minus-end-directed microtubule motors providing the force for flagellar movement [30,31], microtubules and a variety of microtubule-associated proteins [60], kinesins and the trypanosomatid-specific PFR genes (Table 2, Additional files 1 and 3). The genes coding for microtubule-associated proteins were found upregulated in L. major but surprisingly not in L. infantum promastigotes (Table 4). The genes coding for calpains, calcium-dependent cysteine proteinases participating in a variety of cellular processes, including cytoskeletal/membrane attachments and signal transduction pathways [61], were upregulated in L. major and L. infantum promastigotes (Table 2). In contrast, intracellular amastigotes upregulated the expression of the lysosomal cathepsin-L like cysteine proteinases or aminopeptidases (see Additional files 2 and 4).
Species-specific differential gene expression
Comparative analyses of the complete genomes of L. major, L. infantum and L. braziliensis causing distinct human diseases revealed marked sequence conservation and synteny (e.g. more than 99% of genes between the three genomes were syntenic and conservation within coding sequences was 82–94%) [20]. Despite the 20–100 million years of divergence within the Leishmania genus and the broad difference in disease pathologies, only 78 genes were found to be species-specific; 5 L. major-specific, 26 L. infantum-specific and 47 L. braziliensis-specific [20]. Remarkably, our studies revealed that some of these unique species-specific genes, all encoding hypothetical proteins, were also developmentally regulated. For example, LinJ22_V3.0670 only present in L. infantum, or LinJ34_V3.3430, LinJ15_V3.0620, LinJ16_V3.1460, and LinJ31_V3.1180 present in L. infantum but pseudogenes in L. major [20] were expressed preferentially in the amastigote stage (see Additional file 2). The L. major unique genes LmjF04.1020 and LmjF32.2470 were differentially expressed in promastigotes and amastigotes, respectively. The L. major promastigote-regulated genes LmjF10.0185 and LmjF27.0870 are pseudogenes in L. infantum (see Additional file 3). Few of these genes are also present in L. braziliensis although their developmental regulation, if any, is not known. Interestingly, the L. donovani A2 genes (pseudogenes in L. major), the only genes implicated so far in disease tropism [62], are specifically expressed in amastigotes [11]. These data suggest that both parasite genome differences and differential expression of species-specific genes may contribute to species-specific tropism.
The comparative transcriptomic analyses between L. major and L. infantum showed that only 10–12% of the differentially expressed genes in either life stage were common to both species. Commonly upregulated genes may fulfill essential features of the parasite such as metabolism, motility, infectivity and interaction with the host. Consistently, several metabolic genes, the amastigote-specific amastin surface proteins, which may act as receptors or transporters/channels [15] and the promastigote-upregulated dyneins, paraflagellar rod components and calpain-like cysteine peptidases were differentially upregulated in both species. The family of receptor adenylase cyclases (RAC) that play a role in signal transduction was differentially upregulated both in L. major and L. infantum promastigotes (Table 2). This is consistent with the report that rac-A and rac-B genes were expressed preferentially in the insect stage of L. donovani [63]. However, the majority of the differentially expressed genes in either life stage were specific to each Leishmania species. Species-specific differential gene expression may be attributed to a dynamic mode of regulation, which is suitable to species-specific adaptations to different insect vectors and life-cycle features, different target tissues, and distinct disease pathogenesis. Important differences in the stage-regulated gene expression of RNA-binding proteins, amino acid transporters, proteolytic enzymes, and protein kinases were observed between L. major and L. infantum. For example, L. major amastigotes upregulate the expression of important virulence genes encoding the lysosomal cathepsin-L like cysteine proteinases of the papain superfamily [64] whereas L. infantum amastigotes overexpress genes encoding aminopeptidases, which catalyze the removal of N-terminal amino acid residues from peptides and proteins, as well as the subtilisin-like serine peptidase and the hslvu complex proteolytic subunit (see Additional files 2 and 4). Members of the surface antigen proteins 2 (or GP46) are upregulated in L. major promastigotes but not in L. infantum promastigotes (Tables 3, 4 and Additional files 1 and 4).
It is likely that lesion-derived amastigotes and amastigotes isolated from in vitro-infected macrophages display some differences in the expression of genes, especially those that can be modulated by the immune status of the host and/or gene products that contribute together with host factors to the establishment of the disease. However, the majority of modulated genes whose functions are important or essential for the parasite's differentiation into its amastigote form and for the survival of amastigotes within the phagolysosome of macrophages should in principle be modulated similarly in lesion-derived or in vitro-infected macrophages given that amastigotes isolated from THP1 at 4–5 days following infection are fully differentiated and divide within the phagolysosome. The substantial differences that we observed in stage-regulated gene expression not only for amastigotes but also for promastigotes, which were cultured under the same conditions, between L. infantum and L. major may be attributed for the most part to species-specific factors. In line with this possibility are recent observations from our laboratory indicating that gene orthologues between L. major and L. infantum can be regulated in a different manner implying either distinct 3'UTR elements or possibly regulatory factors that are only expressed in one of the species (Müller et al., unpublished). In further agreement with our findings, is a recent interspecies microarray analysis comparing lesion-derived amastigotes of L. major and L. mexicana, two species causing cutaneous leishmaniasis, which demonstrated substantial differences in RNA expression profiles. Indeed, only 15% of the differentially expressed genes in promastigotes were in common between L. major and L. mexicana and there were no amastigote-upregulated genes shared by the two species [65].
We showed here, and others have reported previously [25-29], that a small percentage of genes (7–9%) are regulated at the RNA level throughout the life cycle stages of Leishmania. This percentage of modulated genes is not negligible if one considers that regulation of gene expression in Leishmania occurs at different levels. Several proteomics studies reported a higher amount of genes (12–18%) being regulated in a stage-specific manner at the translational and posttranslational levels [65-67]. It has also been reported that a modest correlation exists between mRNA and protein levels [66]. This modest correlation could be partly explained by the presence of multiple elements within the 3'UTR of Leishmania transcripts, which often have distinct functions on either mRNA stability or translation [46,48,68].
Do stage-regulated transcripts share common regulatory motifs in their 3'UTR?
It is now well established that stage-specific regulation of gene expression in Leishmania is predominantly mediated by elements in the 3'UTR of mRNAs (reviewed in [22,23]). In few cases, specific regions within 3'UTRs have been delineated as cis-elements but their mechanism of action remains for the most part unknown. We recently reported the presence of two classes of distinct short interspersed degenerate retroposons, SIDER1 and SIDER2 that are widely distributed in the 3'UTR of a large number of Leishmania mRNAs (Table 6) [46]. We showed that SIDER1 and SIDER2 have distinct regulatory functions involving either mRNA translational control or RNA degradation, respectively [46-48]. SIDERs were the first conserved regulatory elements found in numerous Leishmania transcripts. However, the distribution of SIDERs among differentially expressed transcripts was not significantly higher from their global distribution among Leishmania transcripts (~25%) (Table 6) [46] (Smith, M. et al., unpublished). SIDER1-mediated regulation occurs at the level of translation and does not necessarily affect mRNA abundance [48]. In the few cases studied, SIDER2 seems to be involved in the degradation of generally short-lived mRNAs. Rapid mRNA turnover may permit the parasite to adapt to the continuously changing physiological needs within its invertebrate and mammalian host, and these temporal changes may not be detectable under our experimental conditions. Hence, SIDERs might participate in more dynamic regulatory processes throughout the Leishmania life cycle in response to specific environmental stimuli, yet to be analyzed. It is likely that additional 3'UTR elements should regulate stability of the differentially expressed transcripts in Leishmania. For example, we have recently identified a U-rich element in the amastin 3'UTR that contributes to the amastin mRNA degradation specifically in promastigotes [68].
Conclusion
In conclusion, whole-genome analyses of inter-stage and inter-species RNA expression profiles between L. major and L. infantum promastigote and amastigote developmental forms highlighted that only a small percentage of Leishmania transcripts are modulated during development and most importantly that Leishmania species causing distinct pathologies express a diverse set of differentially expressed transcripts. These important differences in stage-regulated gene expression between species may be necessary to allow species-specific adaptations to different insect vectors and life-cycle features and could contribute to disease tropism. It is possible that in order to diversify and/or to enlarge their expression profile patterns in response to continuously changing environments throughout development, Leishmania species have adopted a dynamic, multilayered and complex mode of regulating gene expression. The results presented in this study favor this possibility.
Methods
Leishmania strains, cell culture and differentiation
The L. infantum MHOM/MA/67/ITMAP-263 and L. major LV39 MRHO/SU/59/P strains used in this study have been previously described [69,70]. L. infantum and L. major promastigotes were cultured at pH 7.0 and 25°C in SDM-79 medium [71] supplemented with 10% heat-inactivated fetal calf serum (Multicell, Wisent Inc) and 5 μg/ml hemin. L. infantum amastigotes were cultured in a human leukemia monocyte cell line (THP-1 cells) as described previously [72]. THP-1 cells in the log phase of growth were differentiated by incubation for 2 days in medium containing 20 ng of phorbol myristate acetate/ml (Sigma). THP-1 cells were then washed with pre-warmed medium and subsequently infected with stationary-phase L. infantum promastigotes in flat bottom tissue culture flasks (75 cm2) at a parasite/macrophage ratio of 15:1. After two hours, non-internalized parasites were removed by several washes, and infected macrophages were incubated for 4 days. L. major amastigotes were also cultured in THP-1 using the same protocol. L. major lesion amastigotes were obtained from the infected footpad of BALB/c mice 6–8 weeks post-infection as previously described [70].
RNA extraction and cDNA sample preparation
Infected macrophages were incubated with cold HEPES-NaCl-0.0125% SDS for few minutes and neutralized with cold HEPES-NaCl. After centrifugation at 3000 rpm, macrophages were resuspended in cold HEPES-NaCl and, on ice passed 10 times in syringes equipped with a 27G1/2 needle to obtain pure amastigotes free of macrophage material. Amastigotes were washed once in cold HEPES-NaCl and total RNA was prepared using RNeasy Plus (Qiagen) as instructed by the manufacturer. L. major lesion amastigotes were extracted from macrophages using the same procedure as above. Total RNA from promastigotes grown to mid log phase was prepared using RNeasy Plus directly. RNA was treated with Turbo DNAse-free (Ambion). The quality and quantity of RNA were assessed using RNA 6000 Nano Assay Chips (Agilent Technologies). The presence of three distinct ribosomal peaks (18S, 24Sα and 24Sβ) confirmed successful RNA extraction and no detectable macrophage RNA in the L. infantum and L. major amastigote RNA preparations. Complementary DNA (cDNA) was generated from 5 μg of total RNA using a random primer hexamer (GE Healthcare) and aminoallyl-dUTP mix (Sigma) following the protocol for Superscript III (Invitrogen). After 5 hours of incubation at 50°C, the RNA was hydrolyzed with NaOH and EDTA for 15 min at 65°C, then pH was neutralized with HCl. cDNA was purified from unincorporated aminoallyl-dUTP with MinElute PCR Purification columns (Qiagen), phosphate wash buffer (5 mM KPO4 pH 8.0, 80% EtOH) and phosphate elution buffer (4 mM KPO4 pH 8.5).
L. infantum and L. major DNA oligonucleotide full genome microarray design
The recent completion of the sequence of the L. major [19] and L. infantum genomes [20] allowed the generation of multispecies high-density oligonucleotide microarrays. The genome of both species was first compared using BLAST and genes on the same chromosome that had more than 95% homology were grouped together. 70-mer oligonucleotides were designed for each open reading frame of L. infantum and L. major with consistent thermodynamic properties using automated bioinformatic procedures. Probes were initially designed for L. infantum with the added requirement that the region targeted by the probe had optimal homology between both species. For common probes, up to two mismatches were tolerated. In the case that more than 2 mismatches were present in a given gene between L. infantum and L. major, a new probe was designed specifically for L. major (956 probes). The microarray includes in total 8,978 70-mer probes that recognize with no mismatches all L. infantum genes (8,184, GeneDB version 3.0) and also all L. major genes (8,370 genes, GeneDB version 5.1) with the majority of the probes sharing no mismatches and a small percentage of the probes having at most two mismatches (3,580 genome probes are 100% identical between L. infantum and L. major and 3,410 probes have 1–2 mismatches). Also, 622 control probes were included in the microarray for assessing synthesis variability, location of the probe within a given open reading frame and number of mismatches. The probes were synthesised in 384-well plates by Invitrogen (Burlington, Canada). The microarrays were printed on SuperChip (Erie Scientific) using a BioRobotics MicroGrid (Genomic solutions Inc, Ann Arbor, MI). Each probe was printed in duplicate. To assess the ubiquity and specificity of the oligonucleotide probes, the DNA microarray was hybridized in triplicate with genomic DNA isolated from either L. infantum or L. major. Following scanning, both genomic DNA preparations hybridized more than 99% of the oligonucleotides on the array (data not shown). All microarray data will be freely available on the Geo NCBI database in the MIAME format [73]. The series accession number for our manuscript is GSE10407.
Preparation of labeled cDNA and microarray hybridization
Probes for DNA microarray hybridizations were prepared with 10 μg of total RNA for each condition. Purified cDNA from either promastigotes or intracellular amastigotes was dried in speed-vac and resuspended in 0.2 M sodium bicarbonate buffer pH 9.3. Alexa 555 and Alexa 647 dye (Invitrogen) was dissolved in DMSO and incubated in dark with either cDNA for 2 h at room temperature. After coupling, sodium acetate was added and cDNA was purified with MinElute PCR Purification columns (Qiagen) and washed with phosphate washing buffer and phosphate elution buffer. The array was pre-hybridized with 5× complete Denhardt, 30% deionized formamide, 6× SSPE, 0.5% SDS and 0.1 mg/ml ssDNA for 1 h at 42°C and then washed twice for 5 min. at 42°C with 2× SSC, 0.1% SDS, 3 min. at 25°C with 1× SSC, 3 min. at 25°C with 0.2 × SSC and 3 min at 25°C with 0.05 × SSC. Hybridizations were carried out overnight at 42°C (2.5× modified Denhardt, 30% deionized formamide, 6× SSPE, 0.5% SDS, 0.1 mg/ml ssDNA and 0.75 mg/ml yeast tRNA). Multiple biological replicates (4 for L. major and 6 for L. infantum) of all hybridizations were performed to account for sample heterogeneity, variation between slides and variations due to hybridization. To prevent bias by preferential label incorporation into particular sequences, Alexa 555 and Alexa 647 dyes were swapped between the two RNA preparations. Hybridized slides were washed using the pre-hybridization buffer described above.
Fluorescence detection, data processing and statistical analysis
The fluorescence signal intensities of six slides hybridized with L. infantum RNA isolated from promastigotes or amastigotes and four slides hybridized with the L. major RNA (promastigote and amastigote) were measured using the Perkin Elmer ScanArray Express Scanner. GenePix Pro 6.0 image analysis software (Axon Instruments, Union City, California, United States) was employed to measure the fluorescence signal intensities of the array features and local background. Raw data from GenePix were imported in R 2.2.1 for normalization and statistical analyses using the LIMMA (version 2.7.3) package [74-76]. Before processing, probes were flagged according to their hybridization quality on the Leishmania species [43]. Weights were assigned to each array in order to give less weight to arrays of lesser quality [77]. Data were corrected using background subtraction based on convolution of normal and exponential distributions [78]. Intra-array normalization was carried out using the "print-tip loess" method and inter-array normalization was done using the "quantiles of A test" for each array [79]. Statistical analysis was done using linear model fitting and standard errors were moderated using a simple empirical Bayes [74]. Multiple testing corrections were done using the False Discovery Rate (FDR) method with a threshold p-value of 0.05. Only genes statistically significant with an absolute log2 ratio greater than 0.75 were considered as differentially expressed. Further species comparison was performed only on probes that had less than two mismatches when hybridized to either Leishmania species. Thus, 6990 probes could be directly compared between the two species. Gene ontology annotation was analyzed using the AmiGO website [80].
Quantitative Real-Time RT-PCR
Quantitative real-time PCR was carried out on a selected number of genes for validating the microarray experiments (see Table 5 and Additional file 5). Three independent RNA preparations (same used for the microarray experiments) were conducted for each condition. First-strand cDNA was synthesized from 2 μg of total RNA using the Superscript III RNase H Reverse Transcriptase enzyme and random hexamers (GE Healthcare) according to manufacturer's instructions. The resulting cDNA samples were stored at -20°C until use. The primers were designed using Primer Express 2.0 (Applied Biosystems) and their sequence data are the following: × gene: 5-Ztail-N-3; Y gene: 5-Ztail-N-3 (see Additional file 6). Equal amounts of cDNA were run in triplicate and amplified in a 15 μl reaction containing 7.5 μl of 2× universal PCR master mix (Applied Biosystems, Foster City, CA), 10 nM of Z-tailed forward primer, 100 nM of reverse primer, 250 nM of Amplifluor Uniprimer probe (Chemicon, Temecula, California), and 1 μl of cDNA target. Reactions were performed at the Gene Quantification Core Laboratory of the Centre de Génomique de Québec using the Applied Biosystems Prism 7900 Sequence Detector [81]. Three technical and two biological replicates of each reaction were performed, amplification efficiencies were validated and gene expression levels were normalized to constitutively expressed mRNAs encoding the ubiquitin hydrolase gene (LinJ29_V3.2410/LmjF29.2300), 60S ribosomal protein (LinJ18_V3.0630/LmjF18.0620) and a hypothetical protein (LinJ31_V3.0190/LmjF31.0180). Quantification of the relative changes in target gene expression was calculated according to a standard curve.
Authors' contributions
AR performed the microarray experiments, did the mice infections, prepared all Figures and Tables, and participated in the analysis of the data and the writing of the manuscript. FR performed most of the data analysis and participated in the design of the oligonucleotide probes. J-MU established the microarray protocol and helped in the design of the microarray experiments. NM did the macrophage infections. SB helped in the design of the oligonucleotide probes for the construction of the Leishmania full-genome array. MS did the bioinformatics studies on SIDER's distribution among the developmentally regulated transcripts. PR did all the initial bioinformatics work comparing the two Leishmania genomes to design the appropriate oligonucleotide probes for the array. JC and MO participated in the design of the study and reviewed the manuscript. BP participated in the design of the study, coordinated many aspects of the study and wrote the manuscript. All authors have read and approved the final manuscript.
Supplementary Material
Genes differentially expressed in Leishmania infantum promastigotes. This Table lists all the Leishmania infantum genes that are differentially expressed in promastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania infantum intracellular amastigotes. This Table lists all the Leishmania infantum genes that are differentially expressed in intracellular amastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania major promastigotes. This Table lists all the Leishmania major genes that are differentially expressed in promastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania major intracellular amastigotes. This Table lists all the Leishmania major genes that are differentially expressed in lesion-derived amastigotes as determined by DNA microarray studies.
Comparison of expression levels obtained by quantitative real-time PCR between different Leishmania species or experimental models of infection. qRT-PCR analysis was performed on selected differentially expressed genes as determined by microarray experiments. The same RNA used for the microarray analysis was also used for qRT-PCR. (A) Expression values of selected genes in L. major amastigotes isolated either from mouse lesions or from THP1-infected macrophages as determined by qRT-PCR. (B) The qRT-PCR gene expression values of selected genes differentially expressed in L. infantum and/or L. major amastigotes isolated from THP1-infected macrophages. The data are presented as an amastigote to promastigote (Ama/Pro) ratio. Two biological replicates and three technical replicates were included. Error bars denote standard deviations. Values without error bars represent standard deviations lower than 0.05.
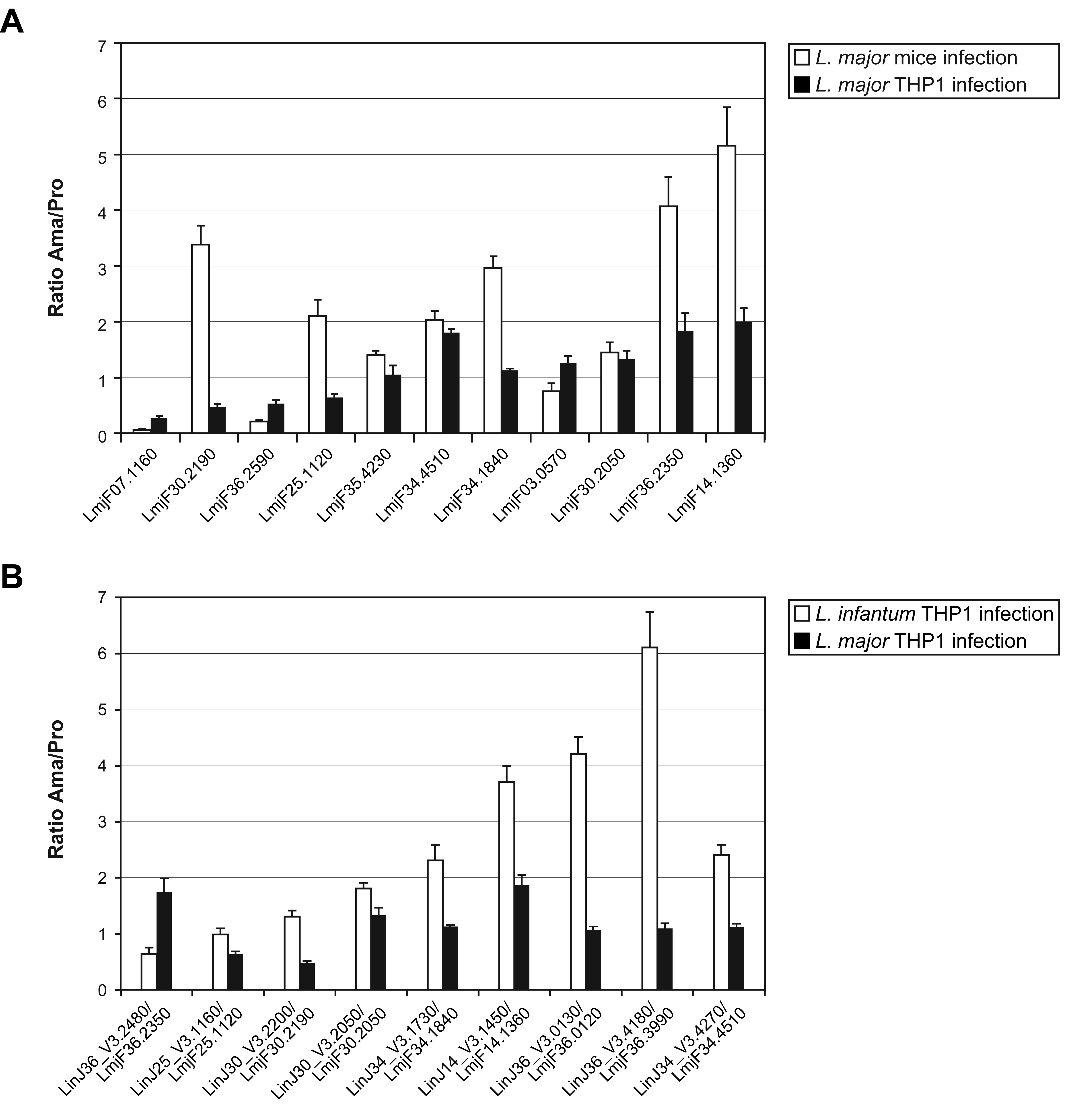{kind=link}
Primers used for quantitative real-time PCR expression analysis. Table lists the sequences of the primers used for quantitative real-time PCR expression analysis to validate DNA microarray studies.
Acknowledgments
Acknowledgements
We thank Gary Hardiman for kindly printing the microarrays, Dr Danielle Légaré for control experiments on the arrays, Dr Éric Madore for helping us with the analysis of the data and Dr Marie Plourde for providing us with amastigote RNA. We appreciated the expertise of the Gene Quantification Facility of the Centre de Génomique de Québec and the Canada Research Chair in Medical Genomics held by Professor Jacques Corbeil. AR is a fellow of Fonds de la Recherche en Santé du Québec. FR and JMU are fellows of the Canadian Institutes of Health Research. M.O. is a Burroughs Wellcome Fund Scholar in Molecular Parasitology and holds the Senior Canada Research Chair in Antimicrobial Resistance. This work was supported by the CIHR operating grant MOP-12182 to BP and the CIHR group grant GR14500 to MO, BP and JC.
Contributor Information
Annie Rochette, Email: annie.rochette@crchul.ulaval.ca.
Frédéric Raymond, Email: frederic.raymond@crchul.ulaval.ca.
Jean-Michel Ubeda, Email: jean-michel.ubeda@crchul.ulaval.ca.
Martin Smith, Email: martin.smith@umontreal.ca.
Nadine Messier, Email: nadine.messier@crchul.ulaval.ca.
Sébastien Boisvert, Email: sebastien.boisvert@usherbrooke.ca.
Philippe Rigault, Email: philippe.rigault@genome.ulaval.ca.
Jacques Corbeil, Email: jacques.corbeil@crchul.ulaval.ca.
Marc Ouellette, Email: marc.ouellette@crchul.ulaval.ca.
Barbara Papadopoulou, Email: barbara.papadopoulou@crchul.ulaval.ca.
References
- Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- WHO Advances in battle against leishmaniasis. TDR News. 1998:2. [PubMed] [Google Scholar]
- Sacks D, Kamhawi S. Molecular aspects of parasite-vector and vector-host interactions in leishmaniasis. Annu Rev Microbiol. 2001;55:453–483. doi: 10.1146/annurev.micro.55.1.453. [DOI] [PubMed] [Google Scholar]
- Zilberstein D, Shapira M. The role of pH and temperature in the development of Leishmania parasites. Annu Rev Microbiol. 1994;48:449–470. doi: 10.1146/annurev.mi.48.100194.002313. [DOI] [PubMed] [Google Scholar]
- Garlapati S, Dahan E, Shapira M. Effect of acidic pH on heat shock gene expression in Leishmania. Mol Biochem Parasitol. 1999;100:95–101. doi: 10.1016/S0166-6851(99)00037-7. [DOI] [PubMed] [Google Scholar]
- Barak E, Amin-Spector S, Gerliak E, Goyard S, Holland N, Zilberstein D. Differentiation of Leishmania donovani in host-free system: analysis of signal perception and response. Mol Biochem Parasitol. 2005;141:99–108. doi: 10.1016/j.molbiopara.2005.02.004. [DOI] [PubMed] [Google Scholar]
- MacFarlane J, Blaxter ML, Bishop RP, Miles MA, Kelly JM. Identification and characterisation of a Leishmania donovani antigen belonging to the 70-kDa heat-shock protein family. Eur J Biochem. 1990;190:377–384. doi: 10.1111/j.1432-1033.1990.tb15586.x. [DOI] [PubMed] [Google Scholar]
- Glaser TA, Moody SF, Handman E, Bacic A, Spithill TW. An antigenically distinct lipophosphoglycan on amastigotes of Leishmania major. Mol Biochem Parasitol. 1991;45:337–344. doi: 10.1016/0166-6851(91)90102-C. [DOI] [PubMed] [Google Scholar]
- Turco SJ, Descoteaux A. The lipophosphoglycan of Leishmania parasites. Annu Rev Microbiol. 1992;46:65–94. doi: 10.1146/annurev.mi.46.100192.000433. [DOI] [PubMed] [Google Scholar]
- McConville MJ, Blackwell JM. Developmental changes in the glycosylated phosphatidylinositols of Leishmania donovani. Characterization of the promastigote and amastigote glycolipids. J Biol Chem. 1991;266:15170–15179. [PubMed] [Google Scholar]
- Charest H, Matlashewski G. Developmental gene expression in Leishmania donovani: differential cloning and analysis of an amastigote-stage-specific gene. Mol Cell Biol. 1994;14:2975–2984. doi: 10.1128/mcb.14.5.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaman M, Aly R, Shapira M. Expression of heat shock protein 83 in Leishmania is regulated post-transcriptionally. Mol Biochem Parasitol. 1994;64:95–110. doi: 10.1016/0166-6851(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Hubel A, Krobitsch S, Horauf A, Clos J. Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol Cell Biol. 1997;17:5987–5995. doi: 10.1128/mcb.17.10.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchmore RJ, Landfear SM. Differential regulation of multiple glucose transporter genes in Leishmania mexicana. J Biol Chem. 1998;273:29118–29126. doi: 10.1074/jbc.273.44.29118. [DOI] [PubMed] [Google Scholar]
- Rochette A, McNicoll F, Girard J, Breton M, Leblanc E, Bergeron MG, Papadopoulou B. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol Biochem Parasitol. 2005;140:205–220. doi: 10.1016/j.molbiopara.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Handman E, Osborn AH, Symons F, van Driel R, Cappai R. The Leishmania promastigote surface antigen 2 complex is differentially expressed during the parasite life cycle. Mol Biochem Parasitol. 1995;74:189–200. doi: 10.1016/0166-6851(95)02500-6. [DOI] [PubMed] [Google Scholar]
- Barr SD, Gedamu L. Cloning and characterization of three differentially expressed peroxidoxin genes from Leishmania chagasi. Evidence for an enzymatic detoxification of hydroxyl radicals. J Biol Chem. 2001;276:34279–34287. doi: 10.1074/jbc.M104406200. [DOI] [PubMed] [Google Scholar]
- Moore LL, Santrich C, LeBowitz JH. Stage-specific expression of the Leishmania mexicana paraflagellar rod protein PFR-2. Mol Biochem Parasitol. 1996;80:125–135. doi: 10.1016/0166-6851(96)02688-6. [DOI] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol Cell. 2003;11:1291–1299. doi: 10.1016/S1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- Clayton C, Shapira M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol. 2007;156:93–101. doi: 10.1016/j.molbiopara.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Haile S, Papadopoulou B. Developmental regulation of gene expression in trypanosomatid parasitic protozoa. Curr Opin Microbiol. 2007;10:569–577. doi: 10.1016/j.mib.2007.10.001. [DOI] [PubMed] [Google Scholar]
- GeneDB website. http://www.genedb.org
- Saxena A, Worthey EA, Yan S, Leland A, Stuart KD, Myler PJ. Evaluation of differential gene expression in Leishmania major Friedlin procyclics and metacyclics using DNA microarray analysis. Mol Biochem Parasitol. 2003;129:103–114. doi: 10.1016/S0166-6851(03)00100-2. [DOI] [PubMed] [Google Scholar]
- Akopyants NS, Matlib RS, Bukanova EN, Smeds MR, Brownstein BH, Stormo GD, Beverley SM. Expression profiling using random genomic DNA microarrays identifies differentially expressed genes associated with three major developmental stages of the protozoan parasite Leishmania major. Mol Biochem Parasitol. 2004;136:71–86. doi: 10.1016/j.molbiopara.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Holzer TR, McMaster WR, Forney JD. Expression profiling by whole-genome interspecies microarray hybridization reveals differential gene expression in procyclic promastigotes, lesion-derived amastigotes, and axenic amastigotes in Leishmania mexicana. Mol Biochem Parasitol. 2006;146:198–218. doi: 10.1016/j.molbiopara.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Leifso K, Cohen-Freue G, Dogra N, Murray A, McMaster WR. Genomic and proteomic expression analysis of Leishmania promastigote and amastigote life stages: the Leishmania genome is constitutively expressed. Mol Biochem Parasitol. 2007;152:35–46. doi: 10.1016/j.molbiopara.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Saxena A, Lahav T, Holland N, Aggarwal G, Anupama A, Huang Y, Volpin H, Myler PJ, Zilberstein D. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol Biochem Parasitol. 2007;152:53–65. doi: 10.1016/j.molbiopara.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston KS, Hill KL. Trypanin, a component of the flagellar Dynein regulatory complex, is essential in bloodstream form African trypanosomes. PLoS Pathog. 2006;2:e101. doi: 10.1371/journal.ppat.0020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron DM, Kabututu ZP, Hill KL. Stuck in reverse: loss of LC1 in Trypanosoma brucei disrupts outer dynein arms and leads to reverse flagellar beat and backward movement. J Cell Sci. 2007;120:1513–1520. doi: 10.1242/jcs.004846. [DOI] [PubMed] [Google Scholar]
- Noll TM, Desponds C, Belli SI, Glaser TA, Fasel NJ. Histone H1 expression varies during the Leishmania major life cycle. Mol Biochem Parasitol. 1997;84:215–227. doi: 10.1016/S0166-6851(96)02801-0. [DOI] [PubMed] [Google Scholar]
- Soto M, Quijada L, Alonso C, Requena JM. Molecular cloning and analysis of expression of the Leishmania infantum histone H4 genes. Mol Biochem Parasitol. 1997;90:439–447. doi: 10.1016/S0166-6851(97)00178-3. [DOI] [PubMed] [Google Scholar]
- D'Orso I, Frasch AC. TcUBP-1, a developmentally regulated U-rich RNA-binding protein involved in selective mRNA destabilization in trypanosomes. J Biol Chem. 2001;276:34801–34809. doi: 10.1074/jbc.M102120200. [DOI] [PubMed] [Google Scholar]
- De Gaudenzi J, Frasch AC, Clayton C. RNA-binding domain proteins in Kinetoplastids: a comparative analysis. Eukaryot Cell. 2005;4:2106–2114. doi: 10.1128/EC.4.12.2106-2114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson DE, Scholtes LD, Myler PJ, Turco SJ, Beverley SM. Genomic organization and expression of the expanded SCG/L/R gene family of Leishmania major: internal clusters and telomeric localization of SCGs mediating species-specific LPG modifications. Mol Biochem Parasitol. 2006;146:231–241. doi: 10.1016/j.molbiopara.2005.12.012. [DOI] [PubMed] [Google Scholar]
- McKean PG, Denny PW, Knuepfer E, Keen JK, Smith DF. Phenotypic changes associated with deletion and overexpression of a stage-regulated gene family in Leishmania. Cell Microbiol. 2001;3:511–523. doi: 10.1046/j.1462-5822.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- Hall MP, Ho CK. Functional characterization of a 48 kDa Trypanosoma brucei cap 2 RNA methyltransferase. Nucleic Acids Res. 2006;34:5594–5602. doi: 10.1093/nar/gkl573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli S, Formenton A, Noll T, Ivens A, Jacquet R, Desponds C, Hofer D, Fasel N. Leishmania major: histone H1 gene expression from the sw3 locus. Exp Parasitol. 1999;91:151–160. doi: 10.1006/expr.1998.4378. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Nelson TN, McMaster WR. Stage-specific expression in Leishmania conferred by 3' untranslated regions of L. major leishmanolysin genes (GP63). Mol Biochem Parasitol. 2001;116:101–104. doi: 10.1016/S0166-6851(01)00307-3. [DOI] [PubMed] [Google Scholar]
- Yao C, Donelson JE, Wilson ME. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol Biochem Parasitol. 2003;132:1–16. doi: 10.1016/S0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Spithill TW, Handman E. The PSA-2 glycoprotein complex of Leishmania major is a glycosylphosphatidylinositol-linked promastigote surface antigen. J Immunol. 1989;143:4221–4226. [PubMed] [Google Scholar]
- Oshlack A, Chabot AE, Smyth GK, Gilad Y. Using DNA microarrays to study gene expression in closely related species. Bioinformatics. 2007;23:1235–1242. doi: 10.1093/bioinformatics/btm111. [DOI] [PubMed] [Google Scholar]
- Singla N, Khuller GK, Vinayak VK. Acid phosphatase activity of promastigotes of Leishmania donovani: a marker of virulence. FEMS Microbiol Lett. 1992;73:221–225. doi: 10.1111/j.1574-6968.1992.tb05322.x. [DOI] [PubMed] [Google Scholar]
- Benzel I, Weise F, Wiese M. Deletion of the gene for the membrane-bound acid phosphatase of Leishmania mexicana. Mol Biochem Parasitol. 2000;111:77–86. doi: 10.1016/S0166-6851(00)00306-6. [DOI] [PubMed] [Google Scholar]
- Bringaud F, Muller M, Cerqueira GC, Smith M, Rochette A, El-Sayed NM, Papadopoulou B, Ghedin E. Members of a large retroposon family are determinants of post-transcriptional gene expression in Leishmania. PLoS Pathog. 2007;3:1291–1307. doi: 10.1371/journal.ppat.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher N, Wu Y, Dumas C, Dube M, Sereno D, Breton M, Papadopoulou B. A common mechanism of stage-regulated gene expression in Leishmania mediated by a conserved 3'-untranslated region element. J Biol Chem. 2002;277:19511–19520. doi: 10.1074/jbc.M200500200. [DOI] [PubMed] [Google Scholar]
- McNicoll F, Muller M, Cloutier S, Boilard N, Rochette A, Dube M, Papadopoulou B. Distinct 3'-untranslated region elements regulate stage-specific mRNA accumulation and translation in Leishmania. J Biol Chem. 2005;280:35238–35246. doi: 10.1074/jbc.M507511200. [DOI] [PubMed] [Google Scholar]
- Smith MA, Blanchette M, Papadopoulou B. Improving the prediction of mRNA extremities in the parasitic protozoan Leishmania. BMC Bioinformatics. 2008;9:158. doi: 10.1186/1471-2105-9-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperdoes FR, Coombs GH. Metabolism of Leishmania: proven and predicted. Trends Parasitol. 2007;23:149–158. doi: 10.1016/j.pt.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Hart DT, Coombs GH. Leishmania mexicana: energy metabolism of amastigotes and promastigotes. Exp Parasitol. 1982;54:397–409. doi: 10.1016/0014-4894(82)90049-2. [DOI] [PubMed] [Google Scholar]
- Bringaud F, Riviere L, Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol. 2006;149:1–9. doi: 10.1016/j.molbiopara.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Rosenzweig D, Smith D, Opperdoes F, Stern S, Olafson RW, Zilberstein D. Retooling Leishmania metabolism: from sand fly gut to human macrophage. Faseb J. 2008;22:590–602. doi: 10.1096/fj.07-9254com. [DOI] [PubMed] [Google Scholar]
- Naderer T, McConville MJ. The Leishmania-macrophage interaction: a metabolic perspective. Cell Microbiol. 2008;10:301–308. doi: 10.1111/j.1462-5822.2007.01096.x. [DOI] [PubMed] [Google Scholar]
- Naderer T, Ellis MA, Sernee MF, De Souza DP, Curtis J, Handman E, McConville MJ. Virulence of Leishmania major in macrophages and mice requires the gluconeogenic enzyme fructose-1,6-bisphosphatase. Proc Natl Acad Sci USA. 2006;103:5502–5507. doi: 10.1073/pnas.0509196103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConville MJ, de Souza D, Saunders E, Likic VA, Naderer T. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends Parasitol. 2007;23:368–375. doi: 10.1016/j.pt.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Mazareb S, Fu ZY, Zilberstein D. Developmental regulation of proline transport in Leishmania donovani. Exp Parasitol. 1999;91:341–348. doi: 10.1006/expr.1998.4391. [DOI] [PubMed] [Google Scholar]
- Geraldo MV, Silber AM, Pereira CA, Uliana SR. Characterisation of a developmentally regulated amino acid transporter gene from Leishmania amazonensis. FEMS Microbiol Lett. 2005;242:275–280. doi: 10.1016/j.femsle.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Besteiro S, Williams RA, Coombs GH, Mottram JC. Protein turnover and differentiation in Leishmania. Int J Parasitol. 2007;37:1063–1075. doi: 10.1016/j.ijpara.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gull K. The cytoskeleton of trypanosomatid parasites. Annu Rev Microbiol. 1999;53:629–655. doi: 10.1146/annurev.micro.53.1.629. [DOI] [PubMed] [Google Scholar]
- Ersfeld K, Barraclough H, Gull K. Evolutionary relationships and protein domain architecture in an expanded calpain superfamily in kinetoplastid parasites. J Mol Evol. 2005;61:742–757. doi: 10.1007/s00239-004-0272-8. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Mendez S, Ghosh A, Myler P, Ivens A, Clos J, Sacks DL, Matlashewski G. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J Biol Chem. 2003;278:35508–35515. doi: 10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- Sanchez MA, Zeoli D, Klamo EM, Kavanaugh MP, Landfear SM. A family of putative receptor-adenylate cyclases from Leishmania donovani. J Biol Chem. 1995;270:17551–17558. doi: 10.1074/jbc.270.29.17551. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Coombs GH, Alexander J. Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol. 2004;7:375–381. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Cohen-Freue G, Holzer TR, Forney JD, McMaster WR. Global gene expression in Leishmania. Int J Parasitol. 2007;37:1077–1086. doi: 10.1016/j.ijpara.2007.04.011. [DOI] [PubMed] [Google Scholar]
- McNicoll F, Drummelsmith J, Muller M, Madore E, Boilard N, Ouellette M, Papadopoulou B. A combined proteomic and transcriptomic approach to the study of stage differentiation in Leishmania infantum. Proteomics. 2006;6:3567–3581. doi: 10.1002/pmic.200500853. [DOI] [PubMed] [Google Scholar]
- Rosenzweig D, Smith D, Myler PJ, Olafson RW, Zilberstein D. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics. 2008 doi: 10.1002/pmic.200701043. [DOI] [PubMed] [Google Scholar]
- Haile S, Dupe A, Papadopoulou B. Deadenylation-independent stage-specific mRNA degradation in Leishmania. Nucleic Acids Res. 2008;36:1634–1644. doi: 10.1093/nar/gkn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereno D, Cavaleyra M, Zemzoumi K, Maquaire S, Ouaissi A, Lemesre JL. Axenically grown amastigotes of Leishmania infantum used as an in vitro model to investigate the pentavalent antimony mode of action. Antimicrob Agents Chemother. 1998;42:3097–3102. doi: 10.1128/aac.42.12.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyombwe A, Olivier M, Harvie P, Bergeron MG, Ouellette M, Papadopoulou B. Protection against Leishmania major challenge infection in mice vaccinated with live recombinant parasites expressing a cytotoxic gene. J Infect Dis. 1998;177:188–195. doi: 10.1086/513821. [DOI] [PubMed] [Google Scholar]
- Brun R, Schonenberger Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop. 1979;36:289–292. [PubMed] [Google Scholar]
- Sereno D, Roy G, Lemesre JL, Papadopoulou B, Ouellette M. DNA transformation of Leishmania infantum axenic amastigotes and their use in drug screening. Antimicrob Agents Chemother. 2001;45:1168–1173. doi: 10.1128/AAC.45.4.1168-1173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Expression Omnibus (GEO) http://www.ncbi.nlm.nih.gov/geo/
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/S1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A, Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hoh J, Broger C, Neeb M, Edington J, Lindpaintner K, Ott J. Statistical methods for analyzing microarray feature data with replications. J Comput Biol. 2003;10:157–169. doi: 10.1089/106652703321825946. [DOI] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centre de génomique du Québec https://genome.ulaval.ca/qrtpcr
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes differentially expressed in Leishmania infantum promastigotes. This Table lists all the Leishmania infantum genes that are differentially expressed in promastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania infantum intracellular amastigotes. This Table lists all the Leishmania infantum genes that are differentially expressed in intracellular amastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania major promastigotes. This Table lists all the Leishmania major genes that are differentially expressed in promastigotes as determined by DNA microarray studies.
Genes differentially expressed in Leishmania major intracellular amastigotes. This Table lists all the Leishmania major genes that are differentially expressed in lesion-derived amastigotes as determined by DNA microarray studies.
Comparison of expression levels obtained by quantitative real-time PCR between different Leishmania species or experimental models of infection. qRT-PCR analysis was performed on selected differentially expressed genes as determined by microarray experiments. The same RNA used for the microarray analysis was also used for qRT-PCR. (A) Expression values of selected genes in L. major amastigotes isolated either from mouse lesions or from THP1-infected macrophages as determined by qRT-PCR. (B) The qRT-PCR gene expression values of selected genes differentially expressed in L. infantum and/or L. major amastigotes isolated from THP1-infected macrophages. The data are presented as an amastigote to promastigote (Ama/Pro) ratio. Two biological replicates and three technical replicates were included. Error bars denote standard deviations. Values without error bars represent standard deviations lower than 0.05.
Primers used for quantitative real-time PCR expression analysis. Table lists the sequences of the primers used for quantitative real-time PCR expression analysis to validate DNA microarray studies.