

Abstract
What evolutionary forces shape genes that contribute to the risk of human disease? Do similar selective pressures act on alleles that underlie simple vs. complex disorders? [1-3]. Answers to these questions will shed light on the origin of human disorders (e.g., [4]), and help to predict the population frequencies of alleles that contribute to disease risk, with important implications for the efficient design of mapping studies [5-7]. As a first step towards addressing them, we created a hand-curated version of the Mendelian Inheritance in Man database (OMIM). We then examined selective pressures on Mendelian disease genes, genes that contribute to complex disease risk and genes known to be essential in mouse, by analyzing patterns of human polymorphism and of divergence between human and rhesus macaque. We find that Mendelian disease genes appear to be under widespread purifying selection, especially when the disease mutations are dominant (rather than recessive). In contrast, the class of genes that influence complex disease risk shows little signs of evolutionary conservation, possibly because this category includes both targets of purifying and positive selection.
Diseases are thought to persist in human populations primarily because of a balance between mutation, genetic drift, and natural selection, with alleles that contribute to disease introduced by mutation, governed in part by random genetic drift, but eventually eliminated from the population by purifying selection [5, 7, 8]. For simple, highly penetrant disorders, purifying selection may be quite strong. For complex diseases, however, individual alleles may contribute little to overall risk and be under only weakly deleterious [9]. Similarly, alleles that cause exclusively late onset Mendelian disorders may not impose an evolutionary fitness cost and thus may be under little or no selection.
Disease susceptibility may also arise, not from a balance between mutation and purifying selection, but as a consequence of adaptation. For example, there is evidence of heterozygote advantage (e.g., at β-globin) and for the fixation of compensatory alleles [10] in genes that cause Mendelian disorders, as well as indications that environmental shifts have led to changes in selection pressures over time. In particular, at a subset of genes associated with complex disease risk, the susceptibility allele is ancestral, and population genetic analyses suggest that the derived, protective allele is selectively advantageous ([3] and references therein). Finally, alleles may be subject to balancing selection if they increase risk of one disease but decrease risk of another, or if there are important interactions between genotype and environment. These considerations raise the possibility that a fraction of loci that underlie contemporary human diseases have been the target of positive, as well as purifying, selection.
To evaluate these hypotheses, the main approach has been to contrast evolutionary rates in genes associated with Mendelian disease phenotypes to all other genes, by using Dn/Ds, the ratio of non-synonymous to synonymous substitutions. Assuming synonymous substitutions are mostly neutral, Dn/Ds reflects the proportion of amino-acid sites in a gene that reach fixation, so are not deleterious. Thus, (1-Dn/Ds) is often thought of as an estimate of the evolutionary constraint acting on a gene (an underestimate if adaptations are frequent), which reflects the extent of purifying selection and, to a lesser extent, its strength.
To date, results of comparisons between disease and “non-disease” genes have been conflicting: Two studies found significantly lower Dn/Ds in genes that cause Mendelian disease than other genes [8, 11], two found significantly higher values [12, 13], and one found no significant difference [14]. These divergent answers may be due to the reliance of most studies on the OMIM database. Although OMIM is the most exhaustive publicly available resource, phenotypic information is sometimes outdated, and is not entered in a standard format, rendering automated searches unreliable (see Suppl. Information 1). A second limitation may be the use, in a subset of papers, of human-rodent comparisons, as it is hard to estimate Dn/Ds reliably for such distantly related species. In addition, many genes classified as non-disease may nevertheless be under strong and widespread purifying selection, reducing the power to detect a difference between categories [8].
To overcome these limitations, we created a hand-curated version of OMIM (hereafter hOMIM), including only highly penetrant diseases caused by a mutation in an autosomal or X-linked gene (see Methods). Since the vast majority of mutations currently known to underlie simple diseases are in exons, we focused on the coding regions, assessing levels of constraints by estimating Dn/Ds between human and rhesus macaque. This Old World Monkey last had a common ancestor with humans over 25 Mya [15], long enough for the comparison to be informative, but short enough for the estimates of Dn/Ds to be reliable, and for the two species to be more likely to share similar pathophysiologies. Finally, we used a classification of essential genes in mice to identify a subset of genes not currently associated with human disease but which are nonetheless likely to be conserved in mammals [16].
Results and Discussion
Analysis of hOMIM genes
We first compared rates of protein evolution among genes in hOMIM (see Methods), with the prediction that, all else being equal, genes in which mutations solely cause late-onset disorders should be less conserved than those in which mutations cause earlier onset disorders. We further expected that if weak purifying selection is common (i.e., if the selection coefficients acting on homozygotes are often in the range -8 < Nes < 0, where Ne is the effective population size) [7, 11], genes in which mutations cause recessive disorders should have higher Dn/Ds than those in which mutations lead to dominant disorders (e.g., Figure 8 in [17]). We therefore tabulated information about the age of onset of the disorder and the mode of inheritance from OMIM entries (see Methods), then assessed whether it predicts the evolution of genes underlying simple disorders. Since the entire coding region is used to test these predictions, a key assumption is that the mode of inheritance and age of onset of disease alleles are predictive of these attributes for other mutations in the same gene.
As expected, most Mendelian disorders with a known genetic basis are early onset, with only a small set manifesting themselves after age 40 (Figure 1). Overall, 45.3% of the disease phenotypes are recessive; the data further suggest that early onset disorders are more likely to be recessive, and late onset disorders dominant, but these findings may also reflect ascertainment bias (e.g., the greater difficulty of mapping loci underlying early onset, dominant disorders).
Figure 1.
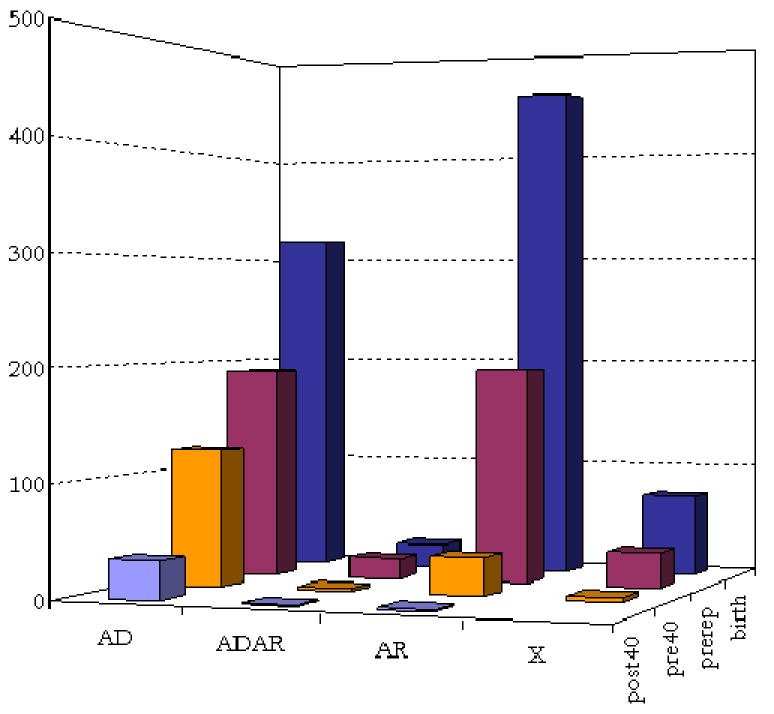
Mode of inheritance and age of onset of disease phenotypes in our hand-curated version of the OMIM database. The data are in Supplementary Materials A.
Considering human-rhesus macaque divergence, we found no evidence that genes in which mutations cause earlier onset disorders have lower Dn/Ds than if the age of onset is late in life (Supplementary Table 1). This could simply reflect lack of power, since we have data on very few genes (14) that cause exclusively late onset disorders; alternatively, mutations in the genes may have pleiotropic effects, or the age of onset may have been earlier in the past [18].
In contrast, we found a highly significant effect of the mode of inheritance on conservation levels of the protein (p ≪ 10-3; see Supplementary Information 1): Dn/Ds values tend to be higher in genes with recessive disease mutations (median = 0.184, n = 452) than those with dominant disease mutations (median = 0.084, n = 294), and intermediate in X-linked genes (median = 0.138, n = 64) (Figure 2; see also [19]). This association could reflect a confounding factor. In particular, the mode of inheritance is known to vary markedly among GO functional categories (e.g., Supplementary Table 2; see Methods for details). However, the mode of inheritance remains a highly significant predictor of Dn/Ds after controlling for this and other possible covariates (Supplementary Table 3).
Figure 2.
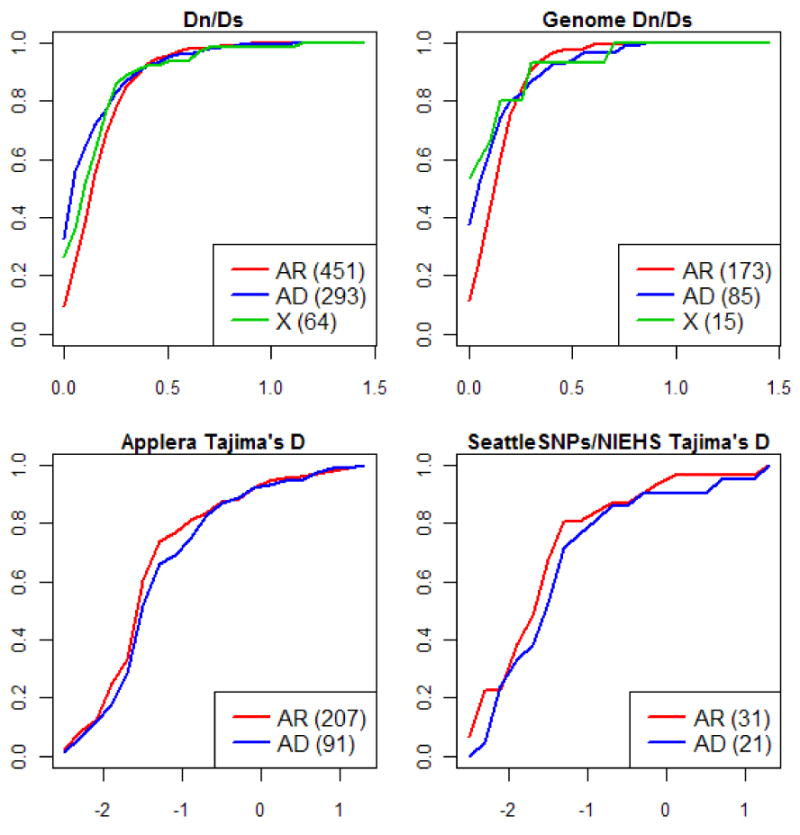
Cumulative distributions of Dn/Ds (for two sets of alignments) and Tajima's D as a function of the mode of inheritance. The value of the statistic is given on the x-axis. AR refers to autosomal recessive and AD to autosomal dominant. In parenthesis are the numbers of genes in each category. The distributions of Dn/Ds for AD and AR categories are significantly different from one another, but the distributions of Tajima's D values are not (see Supplementary Table 1). Tajima's D was calculated for amino-acid variants, using the European population sample; when the African-American sample is used instead, the order of AR and AD is reversed but again the distributions are not significantly different (not shown).
We then combined human polymorphism and human-rhesus macaque divergence data to estimate the fraction of amino-acid sites that are not strongly deleterious, ω. We also estimated the selection coefficient acting on homozygote mutations in disease genes, γ (assuming a fixed selective effect); this value can be thought of as a summary of the pooled polymorphism and divergence data for genes in a given category (see Methods). As shown in Figure 3, there appears to be more widespread and stronger purifying selection on genes associated with dominant rather than recessive disease phenotypes.
Figure 3.
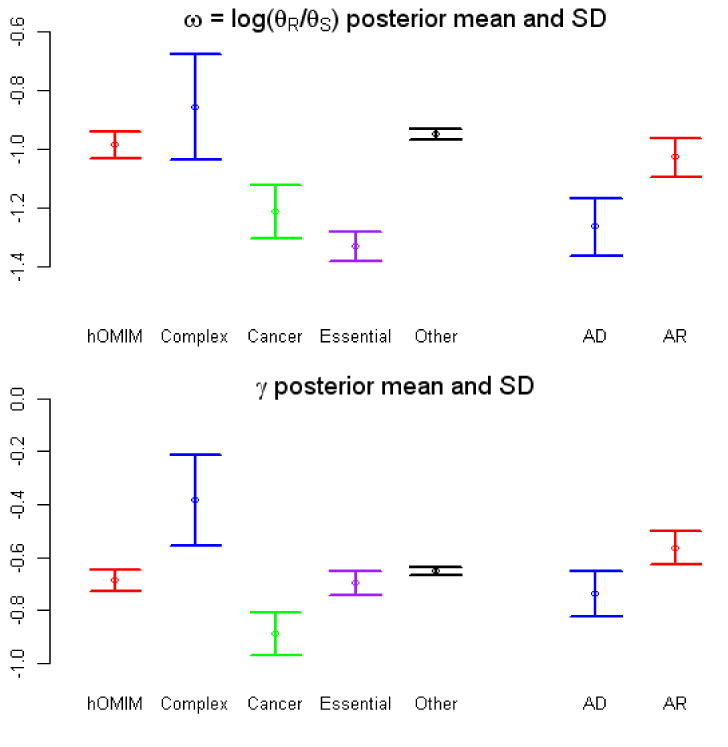
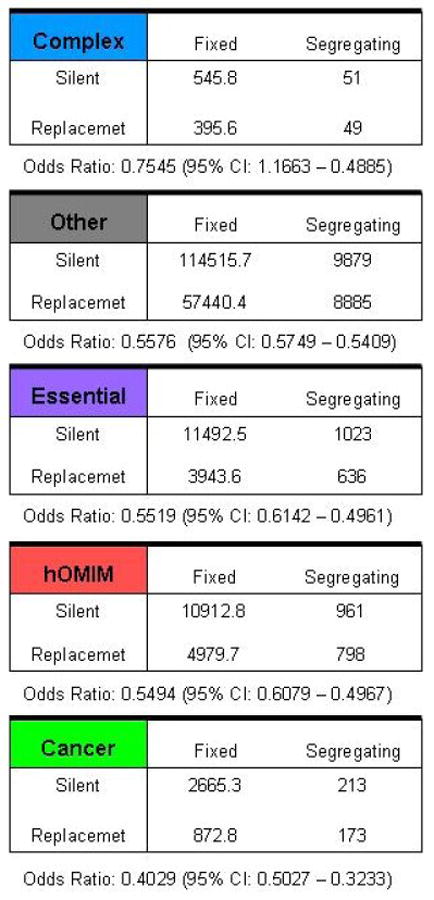
Estimate of two parameters, ω and γ, obtained from pooled polymorphism and divergence data in different categories of genes, including those in hOMIM, those associated with complex disease susceptibility (“complex”), with cancer (“cancer”), for which knock-outs are inviable or sterile in mice (“essential”) and genes in none of the above categories (“other”). Genes in hOMIM are further broken down into two categories, depending on whether mutations cause dominant (“AD”) or recessive (“AR”) disease phenotypes. Shown are the mean and the standard deviation of the posterior distribution estimate for each parameter. The parameter ω=log(θR/θS) can be thought of as the fraction of amino-acid mutations that contribute to polymorphism i.e., are neutral or nearly neutral (θR is the effective mutation rate at replacement sites and θS at synonymous sites), while γ is the selection coefficient acting on mutations in a category of genes. The estimates are obtained by assuming one selection coefficient γ for all mutations within a category; given this unrealistic assumption, the value of the γ estimate is less informative than the ordering for the different categories (see SOM for details). Summaries of the pooled polymorphism and divergence data for genes in each category are given in the last panel (see Methods for details). We note that γ can also be thought of not as a parameter estimate but as a summary of the pooled tables for each category, thereby capturing similar information to the odds ratio (shown below).
Comparison of genes associated with simple vs. complex diseases
Next, we compared conservation levels of genes in hOMIM to those of genes in which mutations are associated with cancer or contribute to other complex disease susceptibility, genes for which knock-outs are inviable or sterile in mouse [16] (hereafter “essential genes”), and genes not known to influence disease risk (see Methods). Comparisons of Dn/Ds suggest that, as a class, proteins that are essential in mouse and those in which mutations are associated with cancer evolve slowest (Figure 4; median Genome Dn/Ds = 0.077 and 0.061, respectively). In turn, the coding regions of hOMIM genes tend to be slightly, but significantly, more slowly evolving than genes not associated with disease (median Genome Dn/Ds = 0.133 vs. 0.139, respectively; see Supplementary Table 1 for p-values).
Figure 4.
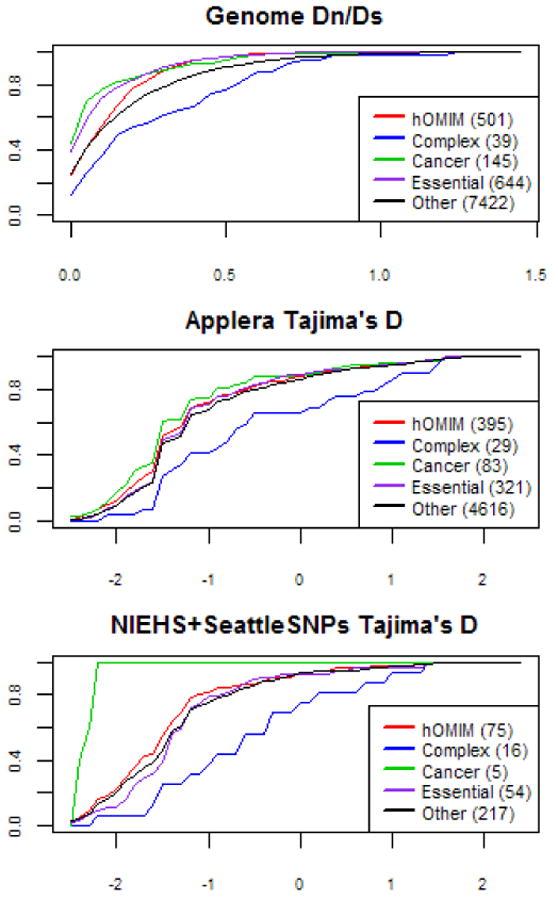
Cumulative distributions of Dn/Ds and Tajima's D for hOMIM, genes associated with complex disease susceptibility (“complex”), in which mutations are associated with cancer (“cancer”), for which knock-outs are inviable or sterile in mice (“essential”) and genes in none of the above categories (“other”). For other details, see legend of Figure 2. The distributions of Dn/Ds are significantly different in all pairwise comparisons (at the 5% level), other than in the comparisons of “essential” genes vs. “cancer” genes and of “other” genes vs. “complex” disease, where significance is marginal (see Supplementary Table 1). The distributions of Tajima's D values in the larger Applera dataset (shown here for the European samples) are significantly different for genes associated with complex diseases vs. either hOMIM or generic genes at the 5% level (see Supplementary Table 1); all other pairwise comparisons are also significant, other than cancer vs. hOMIM, hOMIM vs. essential, and other vs. essential.
The polymorphism data further suggest widespread purifying selection on amino-acid sites in these gene categories (Figure 4). Notably, in all three sets of genes, non-synonymous variants are at significantly lower frequency than synonymous sites (see Supplementary Figure 2). A similar conclusion emerges when combining polymorphism and divergence data to estimate selection parameters ω and γ (Figure 3). Thus, our findings lend further support to the hypothesis that proteins underlying Mendelian disease or associated with human cancers evolve primarily under purifying selection.
While a model of mutation-selection balance was also proposed for genes that influence complex disease risk [7], this group does not show evidence for more conservation than non-disease associated genes. Instead, it tends to have a higher Dn/Ds ratio (median Genome Dn/Ds = 0.203) than hOMIM or other genes, consistent with one of the earlier reports for human-mouse [14]. This difference between genes associated with complex vs. Mendelian diseases is still significant after correction for GO categories, and after exclusion of genes associated with immune response (the median Genome Dn/Ds after exclusion is 0.172; see Methods and Supplementary Figure 1).
In polymorphism data, the allele frequencies of amino-acid variants in genes that influence complex disease susceptibility tend to be higher than in other categories of genes, including genes not associated with disease (Figure 4). Moreover, among genes associated with complex disease susceptibility, allele frequencies do not differ significantly between amino-acid and silent variants (Supplementary Figure 2). These findings do not appear to be explained solely by the ascertainment bias of complex disease gene discovery or the smaller number of genes in this category (see Supplementary Information 1). Together, they suggest that genes associated with complex disease susceptibility tend to be under less pervasive purifying selection than other classes of essential or disease genes. In further support of this conclusion, the estimate of ω is higher for genes associated with complex disease risk than for Mendelian or even for non-disease associated genes, as is the estimate of the selection coefficient, γ (Figure 3).
Why would this be the case? Two (non-mutually exclusive) explanations are that: (i) A substantial fraction of “other genes”, although not known to be essential in mouse or to be associated with human disease, are in fact under widespread and strong purifying selection. In contrast, alleles that contribute exclusively to complex diseases tend to explain only a small proportion of disease risk [9] and to be late onset in their effects, so they may have little fitness consequences. If so, changes in genes associated with complex disease risk may be under very weak, if any, purifying selection. (ii) Genes that influence complex disease susceptibility include loci under widespread purifying selection, but are also enriched for targets of positive selection, thus appearing to be less conserved when considered as a class. For example, if we consider all candidate loci evaluated for evidence of selection by Sabeti et al. [20], there appears to be an enrichment for targets of selection among genes associated with complex disease risk relative to Mendelian disease genes: 8.3% (6 out of 72) genes fall in the empirical 5% tail of the distribution of at least one statistic in at least one population, when only 1.1% of genes in hOMIM do (p = 4.74×10-4, by a two-tailed FET). Complex disease mapping is in its infancy, so that it is too early to distinguish reliably between hypotheses -- especially as the genes that have been found to date are likely an unrepresentative subset (see Methods). Nonetheless, existing data raise the possibility that, while simple disorders are generally well described by models of purifying selection, complex disease susceptibility is tied, at least in part, to evolutionary adaptations.
Methods
Hand-curating OMIM
Our goal was to create a list of all genes that contribute to human diseases with a simple genetic basis. To do so, we used the Online Mendelian Inheritance in Man database (OMIM; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM). OMIM is the most exhaustive, publicly available repository of information about human disease phenotypes. However, it suffers from a number of limitations: for example, entries do not have a standard format, and outdated information is supplemented with new data, rather than replaced. Moreover, while most phenotypic entries are Mendelian, or at least have a simple genetic basis, a non-negligible fraction of entries are clearly complex in etiology (e.g., autism). These features make automated queries highly unreliable.
We therefore decided to create a hand-curated summary of the OMIM database (hereafter referred to as hOMIM), consisting of a list of (gene, phenotype) pairs, together with phenotypic information about the mode of inheritance and age of onset. A description of how the list was constructed is provided in Supplementary Information 1, and the list is available in Supplementary File A. This process yielded a list of 1685 unique (gene, phenotype) pairs for examination, corresponding to 1039 distinct genes. To run our analyses, we excluded phenotypes that were clearly complex or caused by triplet repeat expansions; 1613 (gene, phenotype) pairs remained.
In our analysis of Mendelian disease genes, we also tried using a smaller list of OMIM (gene, phenotype) pairs compiled independently by Jimenez-Sanchez et al. (2001) [21] using slightly different criteria; the qualitative conclusions were the same (results not shown).
List of genes that contribute to complex disease susceptibility
To create a list of genes that influence complex disease susceptibility, we relied on two sources. First, we used compilations in three surveys of association studies [2, 22, 23]. To create a more stringent set of genes, we used only genes for which the associations had been replicated at least once, or where a meta-analysis supported the original association (i.e., bolded entries in Table 2 of [22], as well as entries in Table 2 in ref [23] and Table 1 in ref [2]). Second, we tabulated results from genome-wide association studies of complex disease susceptibility published by June 7, 2007 (see Supplementary Materials B for references). Of the associations reported in these studies, we retained only cases in which the association had been replicated, and where a specific candidate gene had been identified by investigators. From these sources, we found 72 genes associated with complex diseases but are not known to cause Mendelian diseases (i.e., not included on our hand-curated version of OMIM), of which 46 met our more stringent criteria. In our analysis, we considered genes that contribute to both complex disease risk and Mendelian diseases as Mendelian disease genes.
In addition, we analyzed a set of 363 genes in which mutations are associated with cancer susceptibility (http://www.sanger.ac.uk/genetics/CGP/Census/germline_mutation.shtml), as well as a set of genes for which knock-outs were inviable or sterile in mice[16] (downloaded from http://www.umich.edu/∼zhanglab/download/Liao_MBE2006_update/essential.txt). When comparing classes of genes, we classified genes that belong to multiple categories in the following order of priority: hOMIM, complex, cancer, essential, other, so that genes are only in “other” category if not associated with any type of disease and not known to be essential in mouse. We also ran the Dn/Ds analyses excluding the genes that belonged to multiple categories and the results were unchanged (not shown).
GO categories and patho-physiologies
In order to examine the functional annotation of genes, we used the gene ontology (GO) database (http://www.geneontology.org/). Specifically, we retrieved the (level 2) GO assignment of each gene by examining the specific GO terms with which each gene is associated, as determined by EBI (http://www.ebi.ac.uk/). We then located each of these terms on the overall directed acyclic graph (DAG) structure of GO, and traced back to their ancestral terms at this level of annotation. Both the EBI annotations of the genes and the entire DAG structure were downloaded from the database site on September 21st, 2006. In one analysis, we excluded genes associated with immune response, by removing all genes that are associated with the immune system process ontology (GO:0002376) or with any of its subontologies. We also used the pathophysiology classifications of Huang et al. (2004) [13]. This information was available for 99% and 77% of the genes in hOMIM, respectively, for 93% and 28% of genes associated with a complex disorder, and for 96% and 7% of genes in which mutations are associated with cancer. The GO categories for each gene are available in Supplementary Materials C.
Human coding sequences
The Refseq collection of human transcripts was downloaded from ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot on March 18th, 2006. For each gene on our list, we examined all records corresponding to it and selected the longest coding sequence for the gene. In the case of IGKC, Ig kappa chain C region, which does not have a record in refseq, we used the coding sequence in Genbank record BC073791.1.
Estimates of human-rhesus macaque Dn/Ds
For divergence data, we used human-rhesus macaque alignments taken from 10,376 1:1:1 orthologous alignments between human, chimp, and rhesus[24], kindly provided by Adam Seipel at Cornell University. We estimated Dn/Ds for each gene using the PAML package [25], with the default parameters for nuclear DNA. We excluded cases where synonymous divergence was 0, and set Dn/Ds to 0 when non-synonymous divergence was 0. This set of estimates is referred to throughout as the “genome Dn/Ds” values. To map the genes to those on our compilations of disease associations, we used all known gene symbols and aliases from the kgAlias table at the UCSC Genome Database. Genes from the Cornell dataset for which we could not find a symbol were not included in the analysis.
These data only provided alignments for 50% of genes in hOMIM. To increase the number of Mendelian and complex disease genes for which we could estimate Dn/Ds, we also built our own alignments. For this purpose, unassembled sequence of the rhesus macaque genome was downloaded on February 17th, 2006 from http://www.hgsc.bcm.tmc.edu/projects/rmacaque/; for details on how orthology was determined, see Supplementary Information. Each human gene sequence was aligned to its rhesus macaque ortholog using the GAP program [26]. Using the translation of the coding sequence of the human gene, we retained only positions corresponding to whole codons. If an insertion in rhesus macaque sequence occurred within codons, the codons affected by the insertion were removed, as were codons where the rhesus macaque sequence contained a stop codon. We used the PAML package [25] to estimate the Dn/Ds ratio for the resultant pairs of aligned orthologous sequences. Only genes for which the rhesus macaque sequence covered at least 50% of the human sequence were included in the analyses. This process yielded Dn/Ds information for 952 hOMIM genes, 65 genes associated with complex diseases, and 326 genes in which somatic mutations are associated with cancer susceptibility. The Dn/Ds estimates for all genes analyzed are available in Supplementary Materials D.
Human polymorphism data
We analyzed polymorphism data from two resequencing efforts, the NIEHS SNPs (http://egp.gs.washington.edu/) and SeattleSNPs (http://pga.gs.washington.edu/) databases (on August 21, 2006). We analyzed European samples and African (or African-American) samples separately. Sub-Saharan African populations do not appear to have experienced a recent bottleneck, in contrast to European populations (e.g., ref [27]), so that their allele frequencies may be closer to mutation-selection balance. On the other hand, much of the anecdotal evidence for selection on genes associated with complex disease risk is in Europeans (e.g., [28]).
In addition, we analyzed the resequencing polymorphism data in the Applera dataset [11], a genome-wide resequencing effort, considering European-American or African-American samples separately. We also ran the same analyses pooling all population samples, and the qualitative conclusions were unchanged (results not shown). The Applera project also sequenced a chimpanzee to infer the ancestral state, and we used their inference to construct a derived frequency spectrum (see below). We mapped the Applera dataset genes to genes in our lists of Mendelian and complex disease genes as described for the rhesus genome consortium alignments. We used the set of non-synonymous polymorphisms to calculate Tajima's D [29], a summary of the (folded) allele frequency spectrum known to be sensitive to the effects of purifying selection [29]. To do so, we excluded SNPs with small sample sizes (<10 individuals) and more than 10% missing data, as well as genes with 0 non-synonymous polymorphisms. The following formula was used to calculate Tajima's D for each gene: , where , ni is the sample size at site i, and pi is the allele frequency at site i. W was defined following Tajima (1989)[29], as: , where , , S is the number of segregating sites, and nmax is the maximum sample size over all sites. The Tajima's D values are available in Supplementary Materials D.
We also calculated the frequency spectrum for each gene by creating 20 bins of allele frequencies (<5%, 5-10% etc…) and tabulating the number of alleles in each bin. We then created an “average frequency spectrum” for each category (e.g., autosomal dominant) by summing the number in each bin over all genes in that category (effectively concatenating all genes in a given category).
Statistical analyses
To assess whether the distributions of a statistic (Dn/Ds or Tajima's D) differed between two groups of genes (e.g., those in which mutations cause autosomal dominant vs. autosomal recessive disorders), we used a Kolmogorov-Smirnov test. Details are provided in Supplementary Materials 1. To test whether Dn/Ds or Tajima's D predicted the odds of belonging in a given category, we performed logistic regressions using the R function glm with the binomial() parameter (www.r-project.org). A p-value was calculated using the anova function. To examine the selective pressures acting on amino-acid variants, we calculated the mean derived allele frequency for synonymous and for non-synonymous SNPs for each gene. To assess if they differed, a Wilcoxon matched-pairs signed-rank test was performed on the two paired value lists using the wilcox.test() function in R, considering only genes that had both synonymous and non-synonymous SNPs in the sample.
Estimates of γ and ω
We estimated two selection parameters, γ and ω, using a Bayesian method (mkprf) that relies on the entries of a McDonald-Kreitman table [11]. The parameter γ = 2Nes (where Ne is the effective population size) is the scaled selection coefficient acting on homozygous carriers of amino-acid mutations. In turn, ω = log(θR/θS) is a measure of constraint on amino-acid mutations (cf. [30]): θR and θS are estimates of the effective rate of replacement and silent mutations, so that their ratio indicates what fraction of amino-acid mutations can contribute to polymorphism (i.e., is not strongly deleterious).
The mkprf approach uses the number of synonymous and non-synonymous polymorphisms with humans and the number of synonymous and non-synonymous fixed differences between species (here, human and rhesus macaque). Attractive features of the method are that it uses information from polymorphism and divergence jointly, and depends only on the number of polymorphisms, not their frequency, so should be insensitive to possible ascertainment bias effects on the frequency spectrum of genes associated with complex disease. We relied on the polymorphism data from the Applera project, pooling population samples; more details are provided in Supplementary Information 1. Specifically, we summed the entries of the MK tables for all genes within a category (e.g., all genes associated with complex disease susceptibility), excluding X-linked genes (see [11] for details). This approach assumes a fixed selection coefficient across mutations and all genes, effectively averaging over the distribution of selective effects of mutations that contribute to polymorphism or divergence. This highly restrictive assumption makes the absolute value of γ difficult to interpret; however, its ordering across categories is meaningful for a wide variety of distributions of selection coefficients (see Supplementary Materials). Moreover, γ can also be thought of not as a parameter estimate but as a summary of the pooled MK tables for each category, thereby capturing similar information to the odds ratio (see Figure 3). For all genes, we assumed a dominance coefficient h = ½, but we note that, other than in the case of over-dominance (i.e., h>1), this assumption does not affect estimates of the selection coefficients acting on homozygotes [17].
The allele frequency spectrum at genes associated with complex disease
In analyzing the allele frequency at genes associated with complex disorders, it is important to note a number of ascertainment biases. Indeed, genes known to influence complex disease risk have mainly been identified by association studies, so are likely to harbor at least one common allele [5]. We ran resampling analyses to assess the possible effect of this ascertainment bias on Tajima's D and found it to be relatively minor (see Supplementary Information 1), while the effects on Dn/Ds and estimates of γ from the mkprf method are expected to be negligible (see above).
A second consideration is that genes first discovered to influence complex disease risk probably have unusually large effects on the disease phenotype, which implies common alleles yet to be discovered are likely to explain a smaller proportion of the variance. If so, one might predict that the genes yet to be discovered will be under less selection. This said, there may also remain unknown genes associated with complex disease risk that harbor rare alleles of large effect, and are under relatively more conserved than genes identified to date.
Evidence for positive selection at genes associated with disease
Sabeti et al. (2006) [20] considered all genes previously reported to be under positive selection and assessed whether patterns of polymorphism and divergence were unusual relative to background patterns of variation in the genome. For each gene, they reported the percentiles of the distribution of various test statistics designed to detect signatures of selection (their Table S4). We used their criterion, considering a gene to show evidence for selection if it fell in the 5% tail of at least one statistics in at least one of the three populations. This included 6 genes on our list of complex disease genes (out of 72), but only 11 genes in hOMIM (out of 1004). We note that Sabeti et al. predates the publication of one of the best characterized cases of positive selection on a gene associated with complex disease, TCF7L2 [28]. Moreover, the few genes in hOMIM which showed evidence of selection may be unusual, as they include HFE and BRCA1 (which others have considered as associated with complex rather than Mendelian disorders [14]), as well as genes such as G6PD and HBB, which are known to be involved in the resistance to malaria.
Supplementary Material
Acknowledgments
Thanks to G. Coop, N. Cox, A. Di Rienzo, Y. Gilad, C. Ober, G. Sella, M. Stephens, J. D. Wall, W. Wen and especially to J. Pritchard for helpful discussions and/or comments on the manuscript. We are also grateful to the rhesus macaque genome consortium and to A. Siepel in particular for providing a set of human-rhesus alignments, to K. Bullaughey and A. Fledel-Alon for help with bioinformatics, and to M. Groat, L. Stephens, and B. Rashidi for their aid in curating the OMIM database. R. B. is supported by NIH grant GM077959 to Y. Gilad and M.P. by an Alfred P. Sloan fellowship in Computational Molecular Biology and by NIH grant GM79558.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zwick ME, Cutler DJ, Chakravarti A. Patterns of genetic variation in Mendelian and complex traits. Annu Rev Genomics Hum Genet. 2000;1:387–407. doi: 10.1146/annurev.genom.1.1.387. [DOI] [PubMed] [Google Scholar]
- 2.Lohmueller KE, Mauney MM, Reich D, Braverman JM. Variants associated with common disease are not unusually differentiated in frequency across populations. Am J Hum Genet. 2006;78:130–136. doi: 10.1086/499287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Rienzo A. Population genetics models of common diseases. Curr Opin Genet Dev. 2006;16:630–636. doi: 10.1016/j.gde.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Keller MC, Miller G. Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav Brain Sci. 2006;29:385–404. doi: 10.1017/S0140525X06009095. discussion 405-352. [DOI] [PubMed] [Google Scholar]
- 5.Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 6.Cohen JC. Genetic approaches to coronary heart disease. J Am Coll Cardiol. 2006;48:A10–14. [Google Scholar]
- 7.Kryukov GV, Pennachio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. American Journal of Human Genetics. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondrashov FA, Ogurtsov AY, Kondrashov AS. Bioinformatical assay of human gene morbidity. Nucleic Acids Res. 2004;32:1731–1737. doi: 10.1093/nar/gkh330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Consortium, W.T.C.C. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nature Genetics. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondrashov A, Sunyaev S, Kondrashov F. Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci U S A. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, Civello D, Adams MD, Cargill M, Clark AG. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 12.Smith NG, Eyre-Walker A. Human disease genes: patterns and predictions. Gene. 2003;318:169–175. doi: 10.1016/s0378-1119(03)00772-8. [DOI] [PubMed] [Google Scholar]
- 13.Huang H, Winter EE, Wang H, Weinstock KG, Xing H, Goodstadt L, Stenson PD, Cooper DN, Smith D, Alba MM, Ponting CP, Fechtel K. Evolutionary conservation and selection of human disease gene orthologs in the rat and mouse genomes. Genome Biol. 2004;5:R47. doi: 10.1186/gb-2004-5-7-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman MC, Porter A, Czelusniak J, Page SL, Schneider H, Shoshani J, Gunnell G, Groves CP. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol Phylogenet Evol. 1998;9:585–598. doi: 10.1006/mpev.1998.0495. [DOI] [PubMed] [Google Scholar]
- 16.Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- 17.Williamson S, Fledel-Alon A, Bustamante CD. Population genetics of polymorphism and divergence for diploid selection models with arbitrary dominance. Genetics. 2004;168:463–475. doi: 10.1534/genetics.103.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fogel RW. Changes in the disparities in chronic diseases during the course of the 20th century. Perspect Biol Med. 2005;48:S150–165. [PubMed] [Google Scholar]
- 19.Furney SJ, Alba MM, Lopez-Bigas N. Differences in the evolutionary history of disease genes affected by dominant or recessive mutations. BMC Genomics. 2006;7:165. doi: 10.1186/1471-2164-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 21.Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- 22.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 24.Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, Batzer MA, Bustamante CD, Eichler EE, Hahn MW, Hardison RC, Makova KD, Miller W, Milosavljevic A, Palermo RE, Siepel A, Sikela JM, Attaway T, Bell S, Bernard KE, Buhay CJ, Chandrabose MN, Dao M, Davis C, Delehaunty KD, Ding Y, Dinh HH, Dugan-Rocha S, Fulton LA, Gabisi RA, Garner TT, Godfrey J, Hawes AC, Hernandez J, Hines S, Holder M, Hume J, Jhangiani SN, Joshi V, Khan ZM, Kirkness EF, Cree A, Fowler RG, Lee S, Lewis LR, Li Z, Liu YS, Moore SM, Muzny D, Nazareth LV, Ngo DN, Okwuonu GO, Pai G, Parker D, Paul HA, Pfannkoch C, Pohl CS, Rogers YH, Ruiz SJ, Sabo A, Santibanez J, Schneider BW, Smith SM, Sodergren E, Svatek AF, Utterback TR, Vattathil S, Warren W, White CS, Chinwalla AT, Feng Y, Halpern AL, Hillier LW, Huang X, Minx P, Nelson JO, Pepin KH, Qin X, Sutton GG, Venter E, Walenz BP, Wallis JW, Worley KC, Yang SP, Jones SM, Marra MA, Rocchi M, Schein JE, Baertsch R, Clarke L, Csuros M, Glasscock J, Harris RA, Havlak P, Jackson AR, Jiang H, Liu Y, Messina DN, Shen Y, Song HX, Wylie T, Zhang L, Birney E, Han K, Konkel MK, Lee J, Smit AF, Ullmer B, Wang H, Xing J, Burhans R, Cheng Z, Karro JE, Ma J, Raney B, She X, Cox MJ, Demuth JP, Dumas LJ, Han SG, Hopkins J, Karimpour-Fard A, Kim YH, Pollack JR, Vinar T, Addo-Quaye C, Degenhardt J, Denby A, Hubisz MJ, Indap A, Kosiol C, Lahn BT, Lawson HA, Marklein A, Nielsen R, Vallender EJ, Clark AG, Ferguson B, Hernandez RD, Hirani K, Kehrer-Sawatzki H, Kolb J, Patil S, Pu LL, Ren Y, Smith DG, Wheeler DA, Schenck I, Ball EV, Chen R, Cooper DN, Giardine B, Hsu F, Kent WJ, Lesk A, Nelson DL, O'Brien W E, Prufer K, Stenson PD, Wallace JC, Ke H, Liu XM, Wang P, Xiang AP, Yang F, Barber GP, Haussler D, Karolchik D, Kern AD, Kuhn RM, Smith KE, Zwieg AS. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 26.Huang X. On global sequence alignment. Comput Appl Biosci. 1994;10:227–235. doi: 10.1093/bioinformatics/10.3.227. [DOI] [PubMed] [Google Scholar]
- 27.Voight BF, Adams AM, Frisse LA, Qian Y, Hudson RR, Di Rienzo A. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci U S A. 2005;102:18508–18513. doi: 10.1073/pnas.0507325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helgason A, Palsson S, Thorleifsson G, Grant SF, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Schafer H, Faruque M, Doumatey A, Zhou J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Sigurdsson G, Hebebrand J, Pedersen O, Thorsteinsdottir U, Gulcher JR, Kong A, Rotimi C, Stefansson K. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007;39:218–225. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 29.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilad Y, Bustamante CD, Lancet D, Paabo S. Natural selection on the olfactory receptor gene family in humans and chimpanzees. Am J Hum Genet. 2003;73:489–501. doi: 10.1086/378132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.