

Abstract
Cancer is a progressive disease culminating in acquisition of metastatic potential by a subset of evolving tumor cells. Generation of an adequate blood supply in tumors by production of new blood vessels, angiogenesis, is a defining element in this process. Although extensively investigated, the precise molecular events underlying tumor development, cancer progression, and angiogenesis remain unclear. Subtraction hybridization identified a genetic element, progression elevated gene-3 (PEG-3), whose expression directly correlates with cancer progression and acquisition of oncogenic potential by transformed rodent cells. We presently demonstrate that forced expression of PEG-3 in tumorigenic rodent cells, and in human cancer cells, increases their oncogenic potential in nude mice as reflected by a shorter tumor latency time and the production of larger tumors with increased vascularization. Moreover, inhibiting endogenous PEG-3 expression in progressed rodent cancer cells by stable expression of an antisense expression vector extinguishes the progressed cancer phenotype. Cancer aggressiveness of PEG-3 expressing rodent cells correlates directly with increased RNA transcription, elevated mRNA levels, and augmented secretion of vascular endothelial growth factor (VEGF). Furthermore, transient ectopic expression of PEG-3 transcriptionally activates VEGF in transformed rodent and human cancer cells. Taken together these data demonstrate that PEG-3 is a positive regulator of cancer aggressiveness, a process regulated by augmented VEGF production. These studies also support an association between expression of a single nontransforming cancer progression-inducing gene, PEG-3, and the processes of cancer aggressiveness and angiogenesis. In these contexts, PEG-3 may represent an important target molecule for developing cancer therapeutics and inhibitors of angiogenesis.
Genetic changes implicated in cancer development and progression include oncogene activation and tumor suppressor gene inactivation (1–4). Recent studies suggest an additional component to this paradigm, involving genes that are associated with and may directly mediate (progression-elevated genes, PEGen) or suppress (progression-suppressed genes, PSGen) cancer aggressiveness and tumor progression (3, 4). One progression-elevated gene, PEG-3, was identified as a gene displaying elevated expression as a consequence of cancer progression and DNA damage in rodent tumor cells (3). A fundamental question in cancer biology is the mechanism by which these diverse genetic elements interact in mediating tumor development and progression.
An important event in controlling the growth of both primary and metastatic tumors is angiogenesis (5–9). Without neovascularization (formation of new blood vessels), tumors usually do not grow beyond a few cubic millimeters in size (5–7). The formation of new tumor-associated neovascularization is responsible for the increased perfusion of nutrients and oxygen into the tumor mass and the removal of waste products. This process also facilitates entry of tumor cells into the circulatory system, a prerequisite for metastasis. Consistent with this finding, a high degree of tumor vascularization directly correlates with an increase in a tumor's malignant phenotype and inversely correlates with patient survival (10–12). Production of new blood vessels by the developing tumor and distant metastases results from the elaboration of large quantities of angiogenic molecules by both the tumor and host cells (5–9). The balance between positive and negative regulators of this process (8, 9) controls the degree of angiogenesis. These observations emphasize that any genetic modification in a cancer cell that culminates in expansion of tumor growth and metastasis will be inexorably linked to angiogenesis.
Transformation of early passage rat embryo cells by adenovirus type 5 (Ad5) is a progressive process in which morphologically transformed cells temporally acquire new and exhibit further elaboration of existing transformation-related properties (1, 13, 14). Isolating cells after growth in agar, co-expressing additional oncogenes, or reisolating transformed cells after tumor formation in nude mice (13–15) can accelerate this process. Subtraction hybridization of a cDNA library generated from a mutant Ad5- (H5ts125) transformed rat embryo cell clone that forms small, slow-growing, and compact tumors, E11 (1, 13, 14), from a cDNA library produced from a highly aggressive tumorigenic nude mouse tumor-derived E11 clone, E11-NMT (2, 14), resulted in the identification and cloning of PEG-3 (3). Elevated PEG-3 expression occurs in progressed H5ts125-transformed clones and in normal cloned rat embryo fibroblast (CREF) (16) cells displaying a tumorigenic phenotype as a result of expression of diverse acting oncogenes, including Ha-ras, v-src, human papilloma virus type-18-transforming genes, and a specific mutant of Ad5 (H5hr1) (3). When PEG-3 is ectopically expressed in E11 cells, anchorage-independence, an in vitro marker of progression in this model system, is increased (3). These results indicate that PEG-3 can directly contribute to expression of the in vitro transformed phenotype in H5ts125-transformed rat embryo cells.
A number of questions remain concerning the potential role of PEG-3 in regulating the cancer phenotype. These include the biological consequence of elevating PEG-3 expression in normal cells and the in vivo outcome of modifying PEG-3 expression in cancer cells. In the present study, we demonstrate that PEG-3 lacks classical oncogenic potential, but overexpression of this gene in rodent or human tumor cells results in aggressive tumorigenic properties in athymic nude mice. The phenotypic changes induced by overexpression of PEG-3 correlate with an increase in vascular endothelial growth factor (VEGF) production. These findings provide a potential mechanistic framework by which PEG-3 enhances the in vivo cancer phenotype of tumor cells.
Materials and Methods
Cell Lines and Culture Conditions.
CREF is a specific clone of Fischer F2408 rat embryo fibroblast cells (16). E11 is a single-cell clone of H5ts125-transformed Sprague–Dawley secondary rat embryo cells (17). E11-NMT is a subclone of E11 cells derived from a nude mouse tumor (14). E11/PEG-3 S cl 13 and E11-NMT/PEG-3 AS cl 3 are two Zeocin-resistant clones obtained after transfection with pZeoSV/PEG-3 S or pZeoSV/PEG-3 AS, respectively, and selected for growth in 500 μg/ml Zeocin (Invitrogen). The pZeoSV/PEG-3 S and pZeoSV/PEG-3 AS expression constructs were generated by subcloning a full-length PEG-3 cDNA into the pZeoSV mammalian expression vector (Invitrogen) in either the sense or antisense orientation. All cultures were maintained in the logarithmic phase of growth in DMEM supplemented with 5% fetal bovine serum (DMEM-5) at 37°C in a humidified 5% CO2/95% air incubator. Cells resistant to Zeocin were maintained in medium containing 200 μg/ml Zeocin.
Construction and Assaying of PEG-3 Adenovirus (Ad) Vector.
The recombinant replication-defective Ad.PEG-3 S virus was created in two steps and assayed as described previously for producing and assaying Ad.mda-7 antisense and sense viruses (18, 19).
Tumorigenesis Assays.
Growth of E11 and variants in nude mice.
One million of the following lines were injected s.c. into athymic nude mice (NIH Swiss, nu/nu males; Taconic Farms; four animals/group): E11, E11-NMT, E11/PEG-3 S cl 13 (E11 stably transfected with the rat PEG-3 cDNA), and E11-NMT/PEG-3 AS cl 3 (E11-NMT stably transfected with antisense to rat PEG-3). Animals were followed and tumors measured twice weekly with a caliper, and tumor volumes were determined by using the formula π/6 × larger diameter × [smaller diameter]2. Mice were killed when the average tumor volume within a group was >2,000 mm3. Statistical significance between the groups was determined by ANOVA using the computer program sigmastat (Jandel Scientific, San Rafael, CA). A P value of <0.05 was considered significant. Data is presented as tumor volume ± the SD over time. Additionally, one set of two animals/group were killed when the E11/PEG-3 S tumors reached an average tumor volume of 2,000 mm3. These tumors were rapidly excised and fixed in 10% neutral buffered formalin, for use in the immunohistochemical studies.
Effects of rat PEG-3 on the growth of T98G xenografts.
The human glioblastoma multiforme cell line T98G was stably transfected with the rat PEG-3 cDNA or vector using the lipofectamine protocol (18). After drug selection, one million pooled cells from each transfectant (rPEG-3 or vector) were injected s.c. into nude mice (four animals/group). Animals were followed for 4 mo and killed. Tumor volumes were determined as described above.
Effect of Ad.PEG-3 S infection on the growth of DU-145 xenografts.
The human prostate carcinoma cell line DU-145 was infected with the control adenovirus (Ad.Vec) or an adenovirus expressing the rat PEG-3 cDNA (Ad.PEG-3 S). After infection (48 hr), cells were removed with trypsin-EDTA, mixed with Matrigel (1:1, Collaborative Research), and injected s.c. into nude mice. Animals were followed for 1 mo and killed. Tumor volumes were determined as described above.
Immunohistochemistry.
Formalin-fixed tumors were embedded in paraffin, sectioned, and mounted on glass slides (20). Sections were deparaffinized through graded xylene and alcohol washes, and endogenous peroxidase activity was quenched by using methanol-hydrogen peroxide. Sections were blocked with 5% goat serum in PBS, and an mAb to mouse CD31 (PharMingen) was added for 1 hr at room temperature. The sections were washed in PBS, and antibody binding was determined using a Vector ABC kit (Vector Laboratories). After extensive washing, sections were stained with DAB and visualized under the light microscope. In addition to immunostaining, sections also were stained with hematoxylin-eosin to determine tumor morphology and confirm microvessel density.
Nucleic Acid Analyses.
Steady–state levels of PEG-3, VEGF, AdE1A, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA were determined by Northern analysis of total cytoplasmic RNA by using appropriate multiprimed 32P-labeled cloned cDNA probes as described (18, 19). In brief, 10 μg of total RNA from the different cell types were electrophoresed in a 1% agarose gel, transferred to a nylon membrane, and hybridized with the different 32P-labeled cDNA fragments. The membrane was stripped and hybridized with the indicated probes sequentially. Northern blots were washed in a 0.1% SDS, 1× SSC buffer at room temperature for 30 min followed by washing at 42°C for an additional 30 min in the same buffer. Nuclear run-on (in vitro transcription) assays within isolated nuclei were performed as previously described (21, 22). In brief, nuclei from 107 cells were isolated and RNA transcripts previously initiated by RNA polymerase II were allowed to elongate in the presence of 32P-UTP (100 μCi, 3,000 Ci/mmol). The 32P-labeled RNA was extracted with phenol/chloroform, and unincorporated nucleotides were removed by passing the probe through a G-50 sephadex column. Nylon membranes containing 10 μg of the appropriate denatured plasmid DNA gene insert were hybridized with the 32P-labeled RNA. Nylon membranes contained PEG-3, AdElA, basic fibroblast growth factor (bFGF), VEGF, pleiotropin (PTN), midkine (MK), GAPDH, and pBR322 DNA probes. After hybridization, the Nylon membranes were washed and exposed for autoradiography.
Western Blotting Analyses.
The levels of PEG-3 and ACTIN protein were determined by Western blot analysis as described (23). Five million cells were seeded into a 100-mm/tissue culture plate and incubated for 24 hr at 37°C. The medium (DMEM-5) was removed, the cells were washed 3 times with PBS and lysed in RIPC (0.5 M NaCl/0.5% Nonidet P-40/20 mM Tris, pH 8.0/1 mM phenylmethylsulfonyl fluoride). The protein levels were then determined by Western blotting by enhanced chemiluminescence (Amersham, catalog no. RPN 2108) with an anti-PEG-3 peptide-derived rabbit polyclonal antibody and an ACTIN mAb (Santa Cruz Biotechnology). Secreted VEGF protein also was monitored by Western blot analysis (23). Cells were treated as above, except after the DMEM-5 was removed the cells were washed five times with DMEM without fetal bovine serum (DMEM-0). DMEM-0 (5 ml) was then added to each plate, and the cells were incubated for 36 hr at 37°C. The media from the different cell types were harvested and concentrated to 100 μl by using a Centricon filter (Amicon, catalog no. 4211). The levels of secreted VEGF protein were determined by Western blotting by enhanced chemiluminescence with an anti-VEGF antibody (R & D Systems).
VEGF-Promoter Luciferase Assay.
A mouse VEGF promoter (24) (provided by A. P. Adamis, Boston, MA) was subcloned from pGL2BV1.6 vector into the pGL3-basic luciferase reporter vector (Promega E1751). The different cell types were seeded at 5 × 105 cells/35-mm plate 24 hr before transfection. Cells were cotransfected with 5 μg of the pGL3/VEGF vector and 1 μg of the pSV-β-galactosidase (β-gal) vector (Promega E1081) with 10 μl of Lipofectamin (GIBCO). After a 48 hr-incubation at 37°C, cell lysates were prepared and the luciferase activity was determined by using a Luciferase Reporter Gene Assay kit (Boehringer Mannheim). The β-gal activity was determined by using the Galacto-Light Plus kit (Tropix, Bedford, MA). The luciferase data were standardized relative to β-gal activity. To determine the effect of ectopic expression of PEG-3 on VEGF promoter activity in human tumor cells, cultures were infected with 100 plaque-forming units/cell of Ad.PEG-3 S or Ad.Vec (Ad lacking the PEG-3 gene insert); 24 hr later, cells were transfected with pGL3/VEGF and pSV-β-gal vectors, and 48 hr later, luciferase and β-gal activity was determined as described above.
Results
PEG-3 Lacks Transforming or Tumor-Inducing Potential When Expressed in Normal CREF Cells.
Experiments were conducted to determine if PEG-3 is transforming or oncogenic when expressed in normal immortal rat embryo fibroblast cells. CREF cells were transfected with PEG-3 cloned in an expression plasmid containing a Zeocin resistance gene (3). Transfected cells were selected for Zeocin resistance and evaluated for transformed focus formation in cell culture. Additionally, transfected and antibiotic-resistant selected cultures were pooled and injected into nude mice to identify clones with tumorigenic potential. Under these experimental conditions, PEG-3 failed to induce a transformed or oncogenic phenotype (data not shown). In contrast, high molecular weight DNA from various human tumor cell lines (including human prostate carcinoma, breast carcinoma, and glioblastoma multiforme), a primary tumor from a metastasis from a patient with colorectal carcinoma, and various cloned oncogenes, including Ha-ras, Ad E1A, v-src, human papilloma virus type 18, and v-raf, induce morphological transformation in culture and/or an oncogenic phenotype in CREF cells (15, 25, 26). These findings demonstrate that PEG-3 does not function as a classical oncogene, whereas it can modify the phenotype of previously transformed cells (3).
Elevated Expression of PEG-3 in Transformed Rodent and Human Tumor Cells Directly Correlates with an Aggressive Tumor Phenotype in Nude Mice.
To evaluate the oncogenic potential of cells expressing PEG-3, nude mice were injected s.c. with transformed rodent cells endogenously or ectopically expressing PEG-3, E11-NMT, and E11/PEG-3 S, respectively, or expressing reduced levels of PEG-3, E11, and E11-NMT/PEG-3 AS, respectively (Fig. 1). Inoculation of nude mice with E11-NMT or E11/PEG-3 S (three independent clones of E11 cells genetically engineered to express elevated levels of PEG-3) resulted in the formation of aggressive, highly vascularized and rapidly growing tumors (Figs. 1 and 2). In contrast, injection of nude mice with E11 or E11-NMT/PEG-3 AS (two independent clones of E11-NMT cells stably expressing antisense PEG-3 and displaying diminished PEG-3 expression) formed slow growing, compact, and poorly vascularized tumors (Figs. 1 and 2). Fig. 1 shows representative data for a 20-day study using E11/PEG-3 S cl 13 and E11-NMT/PEG-3 AS cl 3; animals were killed at this time because of regulations regarding tumor size. In a separate study, animals injected with E11 and E11-NMT/PEG-3 AS cl 3 cells were maintained for 50 days at which time the final tumor size was still smaller than observed after 20 days in animals injected with E11-NMT or E11/PEG-3 S cl 13 cells (data not shown). Analysis of mRNAs in the different cell strains demonstrated that tumor size and vascularization directly correlate with steady–state PEG-3 mRNA levels, which are elevated in the aggressive tumor-forming cell lines (Fig. 3 and data not shown).
Figure 1.

Effect of PEG-3 expression on oncogenic potential of E11 and E11-NMT cells. Athymic nude mice were injected s.c. with 1 × 106 E11 (○), E11-NMT (□), E11/PEG-3 S cl 13 (⋄) or E11-NMT/PEG-3 AS cl 3 (▵). Mice were evaluated daily until palpable tumors were observed and tumor volumes were measured at day 20.
Figure 2.

Photograph comparing tumor growth of E11 and its derivatives in nude mice. Pictures taken 20 days postinjection with the indicated cell type.
Figure 3.

Gene expression and protein levels as a function of PEG-3 expression. (A) Northern blotting analysis of steady–state PEG-3, VEGF, Ad5 E1A, and GAPDH RNA in E11 and its variants. (B) PEG-3 and ACTIN protein levels were determined by Western blotting using a peptide-derived PEG-3 polyclonal antibody and an ACTIN mAb, respectively, with total lysates from an equal number of cells. To determine VEGF protein secretion, equal numbers of cells were cultured for 36 hr in medium without serum; the medium was collected and concentrated, and VEGF protein levels were determined by Western blotting using a VEGF mAb and the enhanced chemiluminescence approach. Lane designations: 1: E11-NMT; 2: E11-NMT/PEG-3 AS cl 3; 3: E11; and 4: E11/PEG-3 S cl 13.
To determine whether PEG-3 also could regulate cancer aggressiveness in human tumor cells, T98G human glioblastoma multiforme cells were genetically modified by DNA transfection to express the rat PEG-3 coding region. Parental T98G cells did not express PEG-3 or form tumors (after 10 wk) in nude mice, whereas 75% of animals injected with PEG-3 transfected T98G cells produced tumors (Fig. 4). Tumors that developed in the nude mice injected with PEG-3-transfected T98G cells expressed rat PEG-3 (data not shown). Likewise, infection of DU-145 human prostate carcinoma cells with a replication defective type 5 adenovirus expressing PEG-3, Ad.PEG-3 S, 48 hr before injection into nude mice resulted in more rapid tumor development and a statistically significant increase in tumor volume (P < 0.025) (Fig. 4). Moreover, induction of elevated rat PEG-3 expression in diverse human tumors by infection with Ad.PEG-3 S, including carcinomas of the breast (MCF-7 and T47D), cervix (HeLa), and prostate (DU-145), enhanced anchorage independent growth (data not shown). These results document that the rat PEG-3 gene can induce an aggressive oncogenic phenotype and enhance expression of the transformed state in both rodent and human tumor cells.
Figure 4.

Ectopic expression of PEG-3 in T98G and DU-145 tumor cells facilitates tumor formation in vivo in nude mice. (A) Ectopic expression of the rat PEG-3 gene in the low tumorigenic human glioblastoma multiforme cell line T98G induces a progressive tumorigenic phenotype. Pooled cultures of vector or rat PEG-3 cDNA-transfected Zeocin resistant T98G cells were injected into four athymic nude mice/group. Animals were followed weekly until palpable tumors were observed. No tumors developed in vector-transfected T98G cells by 5 mo at which time the experiment was terminated. (B) Ectopic expression of rat PEG-3 in DU-145 cells induces an aggressive oncogenic phenotype. DU-145 cells were infected with 100 plaque-forming units/cell of Ad.PEG-3 S or Ad.Vec (Null, replication incompetent Ad lacking the PEG-3 gene), and 48 hr later the cells were mixed with Matrigel and injected s.c. into athymic nude mice (four animals/group). Animals were monitored as above and tumor volume was determined after 1 mo. *, Statistically significant difference of P < 0.025.
PEG-3 Expression Correlates with Elevated Angiogenesis and Increased VEGF Expression.
On the basis of the morphology of tumors in nude mice induced by PEG-3 expressing rodent tumor cells (highly vascularized and bloody, Fig. 2), we focused on angiogenesis as a possible process underlying the increased in vivo aggressiveness of these tumor cell lines. Analysis of tumors isolated after injection of the different cell strains in nude mice indicated increased formation of blood vessels and elevated CD31 expression, an endothelial cell marker, in PEG-3 expressing transformants (Fig. 5 and data not shown). Similar results were also obtained using mAbs to a second endothelial marker, factor VIII (data not shown). E11-NMT and E11/PEG-3 S cl 13 sections contain multiple microvessels defined by the presence of red blood cells (Fig. 5 C and E) and by CD31 staining (Fig. 5 D and F). Both E11 and E11-NMT/PEG-3 AS cl 3 contain fewer blood microvessels. In addition, E11 and E11-NMT/PEG 3 AS cl 3 tumors appear “ordered” and show fibrotic regions, which are not seen in the E11-NMT or E11/PEG-3 S sections (Fig. 5).
Figure 5.
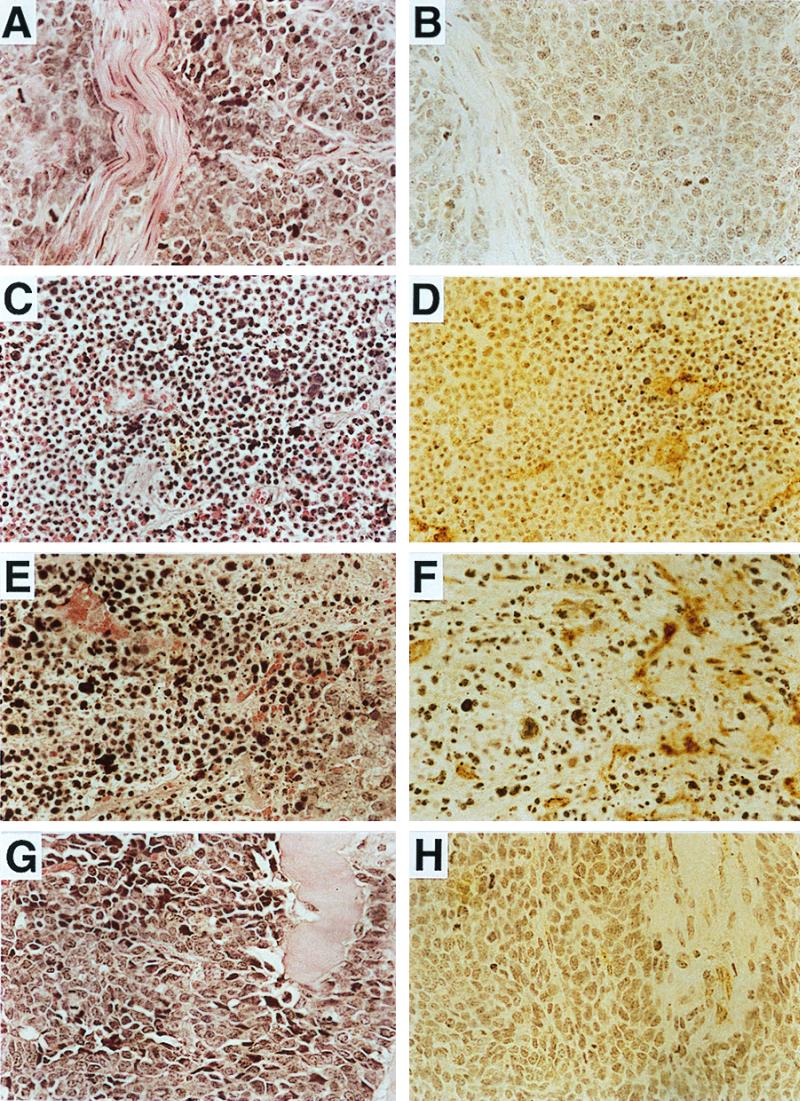
Representative histopathology of formalin-fixed, paraffin-embedded tumors. Serial sections were stained with either hematoxylin-eosin (A, C, E, and G) or immunostained with antibody to the endothelial marker, CD31 (B, D, F, and H). E11 (A and B); E11-NMT (C and D); E11/PEG-3 S cl 13 (E and F); and E11/NMT/PEG-3 AS cl 3 (G and H). (Original magnification, ×200.)
Analysis of mRNAs isolated from the different cell lines indicated higher VEGF mRNA levels in aggressive PEG-3 expressing tumor cells (Fig. 3). In contrast, the mRNA levels of other recognized modulators of angiogenesis, including bFGF (2, 27), PTN (28, 29), and MK (28, 29), did not display consistent alterations as a function of PEG-3 expression (data not shown). Analysis of cell supernatants from the different cell types indicated that the level of secreted VEGF protein was increased in E11-NMT and E11/PEG-3 S cultures vs. E11 and E11-NMT/PEG-3 AS cultures (Fig. 3). In addition, Western blotting analysis of PEG-3 protein levels in the different cell types indicated elevated expression in E11-NMT and E11/PEG-3 S cultures and reduced PEG-3 protein levels in E11 and E11-NMT/PEG-3 AS cells (Fig. 3). These results confirm that PEG-3 expression directly correlates with tumor aggressiveness, and this process associates with elevated expression of the specific angiogenesis regulator VEGF.
Experiments were performed to define the relationship between PEG-3 and VEGF expression. Nuclear run-on assays documented an association between an elevated rate of PEG-3 transcription and increased VEGF transcription in E11-NMT and E11/PEG-3 S cells (Fig. 6). In contrast, although PEG-3 transcription was elevated in E11-NMT/PEG-3 AS cells, VEGF transcription was only marginally higher than in E11 cells. Because antisense inhibition of gene expression can occur at multiple levels, it is possible that the ability of PEG-3 antisense to decrease steady–state PEG-3 mRNA (Fig. 3) occurs at a posttranscriptional level. Confirmation of elevated transcription of the VEGF gene in PEG-3 expressing rodent tumor cells also was obtained by transient expression studies with a VEGF promoter-luciferase reporter construct (Fig. 7). In these experiments, VEGF promoter activity was highest in E11-NMT and E11/PEG-3 cells with decreased activity in E11-NMT/PEG-3 AS cells and the lowest activity in E11 cells. MK transcription also was elevated in E11, E11-NMT and E11-NMT/PEG-3 AS cells, whereas no relationship between PEG-3 and bFGF transcription was apparent and PTN transcription was minimal in all four cell types (Fig. 6). The significance of enhanced MK transcription is unclear because no consistent change in steady–state MK mRNA levels were apparent in the different cell types. These studies document a direct correlation between expression of an aggressive oncogenic phenotype by transformed tumorigenic rodent cells and elevated PEG-3 and VEGF transcription.
Figure 6.

Nuclear run-on analysis of transcription of the PEG-3, Ad5 E1A, bFGF, VEGF, PTN, MK, GAPDH, and pBR322 genes in E11, E11-NMT, E11/PEG-3 S, and E11-NMT/PEG-3 AS cells.
Figure 7.

The activity of a VEGF-promoter-luciferase construct is enhanced in PEG-3 expressing rodent cells. The different cell types were transfected with the pGL3/VEGF vector and a pSV-β-gal vector, and luciferase and β-gal activities were determined. Results are from three independent experiments with triplicate samples per experiment and reflect the average fold-increase in luciferase activity (relative to E11 which represents 1) + SE.
To examine further the relationship between PEG-3 and VEGF, experiments were performed to determine whether transient ectopic expression of PEG-3 in rodent and human tumor cells could elevate VEGF expression. E11 cells were transfected with a PEG-3 expression construct, nuclei were isolated and evaluated for elevated VEGF transcription, total cytoplasmic RNA was isolated, and steady–state VEGF mRNA levels were determined by Northern blotting (Fig. 8 and data not shown). These studies were performed in the presence and absence of cycloheximide to determine if PEG-3 protein was necessary to modify VEGF expression. Elevated VEGF transcription and steady–state RNA levels were apparent in E11 cells 30 hr after transfection with a PEG-3 expression vector, effects that were prevented by cycloheximide (Fig. 8 and data not shown). No changes occurred in the levels of the Ad 5 E1A or GAPDH genes in cycloheximide-treated E11 cells transfected with PEG-3. To determine whether elevating rat PEG-3 expression in human cells could also modify VEGF transcription, a series of human carcinoma cells, including breast (T47D), cervix (HeLa), and prostate (DU-145), were infected with 100 plaque-forming units/cell of Ad.PEG-3 S and transfected with the VEGF promoter-luciferase reporter construct. Under these experimental conditions, the levels of luciferase activity were increased in all of the human carcinoma cell types infected with Ad.PEG-3 S (data not shown). These results suggest that regulation of VEGF expression occurs downstream of PEG-3 expression.
Figure 8.

Transfection of PEG-3 into E11 cells induces steady–state VEGF mRNA. E11 cells were transfected with a pZeoSV/PEG-3 or pZeo SV vector; 4 hr later replicate cultures received cycloheximide, and 30 hr later total RNA was extracted and analyzed by Northern blotting for PEG-3, VEGF, Ad5 E1A, and GAPDH mRNA expression.
Discussion
PEG-3 represents a unique genetic component of the cancer paradigm. It lacks archetypal oncogenic potential but can directly facilitate expression of an aggressive cancer phenotype in both rodent and human tumor cells. The present study provides evidence for a relationship between PEG-3 expression and transcriptional activation of the angiogenic-regulating molecule VEGF. In this context, PEG-3 may directly contribute to cancer aggressiveness by facilitating blood flow to the tumor resulting in increased growth and access to the circulatory system by potentially metastatic tumor cells. Although one level by which PEG-3 regulates VEGF expression involves enhanced transcriptional control, further studies are required to determine whether PEG-3 can also affect the processing of VEGF mRNA or its translation or secretion from tumor cells. In addition, because elevated PEG-3 expression also augments anchorage independent growth in tumor cells, increased cancer aggressiveness also may reflect additional changes in cancer cell physiology resulting in increased tumor growth in vivo. A recently developed gene cloning approach, reciprocal subtraction differential RNA display (RSDD) (4), resulted in the identification of several novel genes displaying either progression elevated or progression suppressed gene expression as a function of transformation progression in the E11 to E11-NMT model. The role of these genes in the progression process and how they interact with PEG-3 in regulating cancer progression are interesting and relevant questions currently under investigation.
The mechanism by which PEG-3 modifies the cancer phenotype remains to be elucidated. PEG-3 is inducible in normal rat embryo cells by DNA damage and expression is elevated as a function of both cancer progression and oncogene-mediated transformation (3). The ability of PEG-3 expression to be induced in CREF cells by DNA damage is shared with two genes with significant homology to PEG-3, a growth arrest and DNA damage inducible hamster gene, gadd34 (30), and its homologous differentiation regulated murine gene, MyD116 (31). However, in contrast to these structurally related genes, only PEG-3 expression coincides with the progression phenotype (3). Additional support for a relationship between PEG-3 expression, cancer progression, and angiogenesis is provided by antisense PEG-3 studies. Expression of PEG-3 antisense in E11-NMT cells suppresses PEG-3 mRNA and protein levels, inhibits the progression phenotype, and reduces VEGF steady–state mRNA and protein secretion. In this context, PEG-3 expression may serve as a sensitive biosensor for identifying therapeutic agents and chemically generated small molecules that can function as novel inhibitors of cancer progression and angiogenesis.
Diverse acting oncogenes, including Ha-ras, v-src, H5hr1, v-raf, and HPV-18, induce a tumorigenic phenotype in CREF cells, which correlates with elevated expression of PEG-3. Because the mode of action of these diverse acting oncogenes is different, PEG-3 may represent a common downstream target gene required for the activation of oncogenic potential by cancer cells. Although its role in the multistep carcinogenic process requires further clarification, PEG-3 may function as a “gatekeeper” that regulates the final cascade of gene expression changes required for a cancer cell to progress to a more aggressiveness state (3). Identification of these cancer regulatory molecules should provide insights into the complex controls and processes underlying cancer evolution.
Although the mechanism by which PEG-3 expression positively influences the carcinogenic process remains to be defined, PEG-3 may potentiate this process by eliciting a constitutive DNA damage stress response that facilitates genomic instability and cancer progression, by inducing an aggressive cancer phenotype and angiogenesis. Defining the mode of action of PEG-3 is clearly a worthwhile endeavor and offers promise for identifying relevant target molecules and pathways that may be exploitable for cancer diagnosis and therapy.
Acknowledgments
The present study was supported in part by National Institutes of Health Grants CA35675, CA73264, and GM31452, the Chernow Endowment, and the Sam Waxman Cancer Foundation. P.B.F. is the Michael and Stella Chernow Urological Cancer Research Scientist.
Abbreviations
- PEG-3
progression elevated gene-3
- VEGF
vascular endothelial growth factor
- β-gal
β-galactosidase
- Ad5
adenovirus type 5
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- CREF
cloned rat embryo fibroblast
- bFGF
basic fibroblast growth factor
- MK
midkine
- PTN
pleiotropin
- NMT
nude mouse tumor
References
- 1.Fisher P B. In: Tumor Promotion and Cocarcinogenesis In Vitro: Mechanisms of Tumor Promotion. Slaga T J, editor. Boca Raton, FL: CRC; 1984. pp. 57–123. [Google Scholar]
- 2.Fidler I J. Cancer Res. 1990;50:6130–6138. [PubMed] [Google Scholar]
- 3.Su Z-z, Shi Y, Fisher P B. Proc Natl Acad Sci USA. 1997;94:9125–9130. doi: 10.1073/pnas.94.17.9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang D-c, LaFrance R, Su Z-z, Fisher P B. Proc Natl Acad Sci USA. 1998;95:13788–13793. doi: 10.1073/pnas.95.23.13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkman J. J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 6.Folkman J. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 7.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 8.Liotta L A, Stetler-Stevenson W G. Cancer Res. 1991;51:5054s–5059s. [PubMed] [Google Scholar]
- 9.Kumar R, Fidler I J. In Vivo. 1998;18:27–34. [PubMed] [Google Scholar]
- 10.Weidner N, Folkman J, Pozza F, Bevilacqua P, Alfred E N, Moore D H, Meli S, Gaspanni G. J Natl Cancer Inst. 1992;84:1875–1877. doi: 10.1093/jnci/84.24.1875. [DOI] [PubMed] [Google Scholar]
- 11.Weidner N, Carroll P R, Flax J, Blumenfeld W, Folkman J. Am J Pathol. 1993;143:401–409. [PMC free article] [PubMed] [Google Scholar]
- 12.Weidner N, Folkman J. In: Important Advances in Oncology. DeVita V T, Hellman S, Rosenberg S A, editors. Philadelphia: Lippincott; 1996. pp. 167–190. [Google Scholar]
- 13.Fisher P B, Bozzone J H, Weinstein I B. Cell. 1979;18:695–705. doi: 10.1016/0092-8674(79)90124-7. [DOI] [PubMed] [Google Scholar]
- 14.Babiss L E, Zimmer S G, Fisher P B. Science. 1985;228:1099–1101. doi: 10.1126/science.2581317. [DOI] [PubMed] [Google Scholar]
- 15.Fisher P B, Babiss L E, Weinstein I B, Ginsberg H S. Proc Natl Acad Sci USA. 1982;79:3527–3531. doi: 10.1073/pnas.79.11.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy P G, Su Z-z, Fisher P B. In: Chromosome and Genetic Analysis, Methods in Molecular Genetics. Adolph K W, editor. Vol. 1. Orlando, FL: Academic; 1993. pp. 68–102. [Google Scholar]
- 17.Fisher P B, Eisenberg D, Weinstein I B, Ginsberg H S. Proc Natl Acad Sci USA. 1978;75:2311–2314. doi: 10.1073/pnas.75.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang H, Su Z-z, Lin J J, Goldstein N I, Young C S H, Fisher P B. Proc Natl Acad Sci USA. 1996;93:9160–9165. doi: 10.1073/pnas.93.17.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su Z-z, Madireddi M T, Lin J J, Young C S H, Kitada S, Reed J C, Goldstein N I, Fisher P B. Proc Natl Acad Sci USA. 1998;95:14400–14405. doi: 10.1073/pnas.95.24.14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su Z-z, Lin J, Shen R, Fisher P A, Goldstein N I, Fisher P B. Proc Natl Acad Sci USA. 1996;93:7252–7257. doi: 10.1073/pnas.93.14.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Z-z, Austin V A, Zimmer S G, Fisher P B. Oncogene. 1993;8:1211–1219. [PubMed] [Google Scholar]
- 22.Jiang H, Waxman S, Fisher P B. Mol Cell Different. 1993;1:197–214. [Google Scholar]
- 23.Su Z-z, Yemul S, Estabrook A, Zimmer S G, Friedman R M, Fisher P B. Int J Oncol. 1995;7:1279–1284. doi: 10.3892/ijo.7.6.1279. [DOI] [PubMed] [Google Scholar]
- 24.Shima D T, Kuroki M, Deutsch U, Ng Y-s, Adamis A P, D'Amore P A. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- 25.Su Z-z, Olsson C A, Zimmer S G, Fisher P B. Anticancer Res. 1992;12:297–304. [PubMed] [Google Scholar]
- 26.Lin J, Su Z-z, Grunberger D, Zimmer S G, Fisher P B. Int J Oncol. 1994;5:5–15. doi: 10.3892/ijo.5.1.5. [DOI] [PubMed] [Google Scholar]
- 27.Folkman J, Klagsbrun M. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- 28.Bohlen P, Kovesdi I. Prog Growth Factor Res. 1991;3:143–157. doi: 10.1016/s0955-2235(05)80005-5. [DOI] [PubMed] [Google Scholar]
- 29.Garver R I, Radford D M, Doris-Kellen H, Wick M R, Milner P G. Cancer (Phila) 1994;74:1584–1590. doi: 10.1002/1097-0142(19940901)74:5<1584::aid-cncr2820740514>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 30.Fornace A J, Jr, Alamo I, Jr, Hollander M C. Proc Natl Acad Sci USA. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lord K A, Hoffman-Liebermann B, Liebermann D A. Nucleic Acids Res. 1990;18:2823. doi: 10.1093/nar/18.9.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]