
Figure 3.
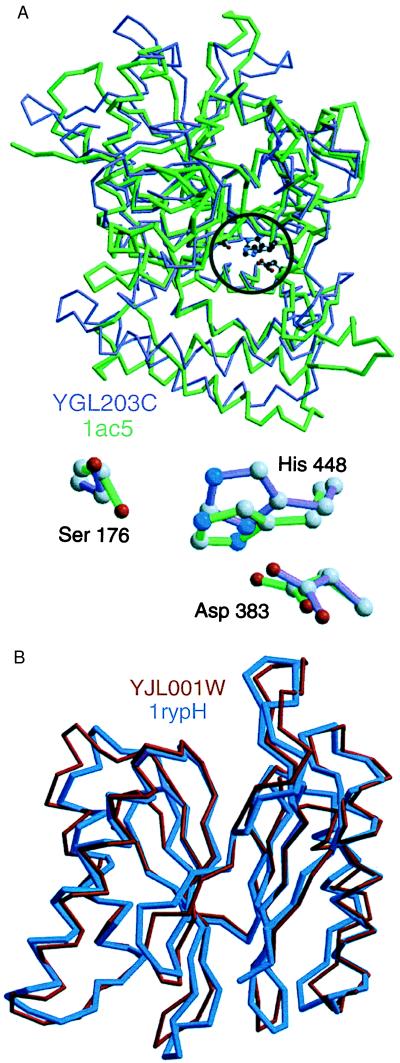
Sample models calculated before the crystallographic structures have been deposited to PDB. (A) A model for the yeast prohormone-processing carboxypeptidase (YGL203C, violet) is compared with its actual crystallographic structure (1ac5, green) (38). The model was constructed based on the crystal structure of the yeast serine carboxypeptidase (1cpy) with which it shares only 25% sequence identity. Although the overall structural overlap of the model and the actual structure is only 63%, the active site (Inset) and the neighboring residues have been modeled with useful accuracy; for example, it is possible to use the model to plan site-directed mutagenesis experiments for assessing residues critical for catalysis and binding specificity. The model also illustrates that the functionally important regions of the molecule tend to be modeled more accurately than the rest of the protein (Fig. 1B) because they are frequently more conserved in evolution than the rest of the fold. (B) A model for the yeast multi-catalytic protease (YJL001W, red) is compared with its actual crystallographic structure (1rypH) (30). Despite a low sequence identity of 24% to the template structure (1pmaB), the model overlaps with the actual x-ray structure in 92% of the residues (point δ in Fig. 1B). It was possible to predict that this particular model was unusually accurate given its sequence similarity to the template because it had a favorable Z-score of −8.3 and an energy profile with only one positive peak (19). The YJL001W subunit is part of the 20S proteasome, a highly ordered ring-shaped structure consisting of 14 similar subunits, all of which have been modeled in this study. The models are sufficiently accurate for use with protein–protein docking programs, which in turn are likely to predict correctly at least some of the interface residues between the subunits (17).