
Figure 4.
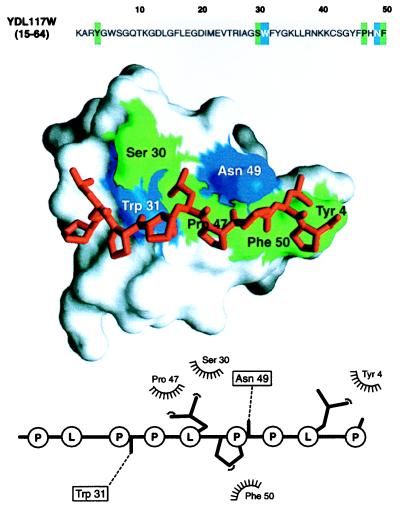
Modeling a putative interaction of a predicted YDL117W SH3 domain with a proline-rich peptide. A segment in the yeast ORF YDL117W sequence (Top) was predicted to be remotely related to the SH3 domains, many of which have known 3D structure (Table 1). The automated prediction was possible because of the sensitivity afforded by evaluating a 3D model implied by the match. The 3D model of the SH3 domain in turn allowed us to address the biochemical function of YDL117W by calculating a 3D model of a complex between the predicted SH3 domain and a putative ligand, a proline-rich peptide (Middle). Inspection of the YDL117W sequence revealed that there is a proline-rich segment downstream from the putative SH3 domain (PLPPLPPLP, positions 212–220). Because this peptide contains the signature PXXP sequence typical of the SH3 binding peptides (39), it was the ligand chosen for the modeling of the complex; both inter- and intramolecular interactions between SH3 domains and Pro-rich peptides already have been documented (39). A model of the complex was obtained by the same comparative method as the model of the SH3 domain (15), relying on the crystallographic structure of the complex between the FYN SH3 domain and its peptide ligand (PPAYPPPPVP) (40). The predicted SH3 domain is shown in the surface representation (41), with the ball-and-stick model of the peptide (red) lying in the binding site. The SH3 residues making hydrophobic contacts and hydrogen bonds to the ligand peptide are colored in green and blue, respectively. The bottom panel shows a schematic representation of the SH3-peptide interaction (42). The peptide atoms that interact with the SH3 residues are shown as filled spheres, hydrogen bonds are represented by dashed lines, and hydrophobic interactions are indicated by the spiked semicircles. This model facilitates designing experiments such as site-directed mutagenesis for maping of functionally important residues on the SH3 domain and its ligand. This should be compared to the starting point at which no functional information about this ORF or about the proteins related to it was known. More generally, the wealth of information in the bottom two panels relative to the top, sequence-only panel is a case in point for the utility of structural models in planning biological experiments (see also text). For the many proteins whose structures have not been determined by experiment, maximal structural information is obtained by both (i) establishing a match to a known protein structure and (ii) calculating an all-atom 3D model based on that match by using the methods described in this paper.