

Abstract
RNA helicase A (RHA) is the human homologue of the Drosophila maleless protein, an essential factor for the development of male flies. Recently, it was shown that RHA cooperates with the cAMP-responsive element in mediating the cAMP-dependent transcriptional activation of a number of genes. Due to the participation of cAMP as a second messenger in a number of signaling pathways, we examined the function of RHA during mammalian embryogenesis. To examine the role(s) of RHA in mammalian development, RHA knockout mice were generated by homologous recombination. Homozygosity for the mutant RHA allele led to early embryonic lethality. Histological analysis, combined with terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) reactions of RHA-null embryos, revealed marked apoptotic cell death specifically in embryonic ectodermal cells during gastrulation. RNA in situ analyses of the expression of HNF-3β and Brachyury, two molecular markers for gastrulation, showed that RHA-null embryos at days 7.5 and 8.5 expressed both HNF-3β and Brachyury in a pattern similar to those of pre- and early streak stages of embryos, respectively. These observations indicate that RHA is necessary for early embryonic development and suggest the requirement of RHA for the survival and differentiation of embryonic ectoderm.
RNA helicase A (RHA) is the only known nuclear enzyme that catalyzes the displacement of both double-stranded RNA and DNA (1, 2). The sequence of the cDNA of RHA revealed that it belongs to the Asp-Glu-Ala-His (DEAH) family of ATPase/helicase proteins and that it is the human homologue of the Drosophila maleless protein (MLE) (3). Bovine nuclear DNA helicase II is also an RHA homologue, exhibiting identical biochemical properties (4, 5).
The evolutionary conservation of the sequence and biochemical properties of RHA and its homologues (2–6) suggest that their biological role may be also conserved. Because RHA can function as a helicase with both RNA and DNA, it may participate in various nuclear transactions, including transcription and post-transcriptional processes. Recently, it has been reported that RHA interacts with the cAMP-responsive element binding protein (CREB)-binding protein (CBP), and that transcriptional activation in response to cAMP requires both RNA helicase A and CBP (7). The observation that a point mutation (Lys to Asn) introduced into the conserved ATP binding motif (Gly-Lys-Thr) of RHA resulted in a reduction in the level of transcription (7) suggests that the ATP binding and/or ATP hydrolysis activities of RHA are required for efficient cAMP-mediated transcriptional activation.
In Drosophila, dosage compensation is achieved by increasing the transcriptional activity of X-linked genes in males (8). Among the five protein factors known to be essential for dosage compensation in Drosophila (MLE; male-specific lethal-1, -2, and -3; and males-absent on the first). MLE is the only protein whose biochemical properties have been well defined (3). Both in vitro and in vivo studies employing site-directed mutagenesis established that NTPase/helicase activities not only are associated with MLE but also are critical for dosage compensation in male flies.
In mammals, dosage compensation is achieved by suppressing the transcription of genes located on one of the two X chromosomes in females (XX) (9). At present, XIST/Xist is the only factor known to be involved in this X-inactivation process (10–12). The apparent differences in the pathway of dosage compensation between Drosophila and humans make it unlikely that RHA plays a sex-specific role in mammalian development. In light of the essential role of NTPase/helicase activities for transcriptional activation (3, 7), it is more likely that the molecular basis by which RHA and MLE achieve transcriptional activation is mechanistically conserved. Because both RHA and CBP/p300 are required for transcriptional activation in response to cAMP and a number of genes are known to be regulated by the cAMP signaling pathway during development and differentiation (13, 14), we examined the role of RHA in mammalian development. Our observations demonstrate that RHA is essential for embryonic development in mice and plays a critical role in the normal progression of gastrulation.
MATERIALS AND METHODS
Construction of a Targeting Vector.
The 7.6-kb KpnI–KpnI upstream region of the RHA gene (genomic region 7.2 to 14.8 kb) (6) was subcloned into pBluescript II SK(−) and the Neo gene, the positive selection marker, was inserted in the middle of exon II. The tk gene, the negative selection marker, was introduced into a specific KpnI site present between exons IV and V of the RHA gene. A more detailed procedure is available upon request. The targeting vector was linearized with HindIII, which cleaved the 5′ end of the tk gene in pBluescript II SK(−), and the product was electroporated into CJ7 embryonic stem (ES) cells as described (34). ES clones were selected in culture media containing G418 (250 μg/ml) and ganciclovir (2.5 μM) for 10 days, and subjected to Southern blot analysis by using a N-terminal cDNA probe prepared with random primers and the 593-bp XbaI fragment of mouse RHA cDNA as template as described (6, 34). Homologous recombination was observed at a frequency of about 1/100 G418/ganciclovir-resistant clones.
Genotyping.
Genotyping of liveborn offspring and embryos by Southern blot analysis was performed by using the probe B prepared with the 1.3-kb KpnI–XbaI fragment of the RHA gene as template (genomic region 14.8–16.1 kb) (6). PCR analysis was carried out by using two synthetic primers (5′ primer of 5′-GAAGACACCTGAATCATGGGTGA-3′ and 3′ primer of 5′-CTTTAAACCAGACGAACTTCACAAG-3′), which yielded an 180-bp PCR product containing the last 126 bp of exon II and the first 54 bp of intron II as described (6). To prepare genomic DNAs from embryonic day 10.5 (E10.5) embryos, embryos were dissected free of maternal tissue and parietal yolk sac with fine forceps. For Southern blot analysis, the visceral yolk sac from phenotypically normal embryos was used, whereas the entire abnormal embryo was used due to the limitation of available materials (arrow in Fig. 2).
Figure 2.
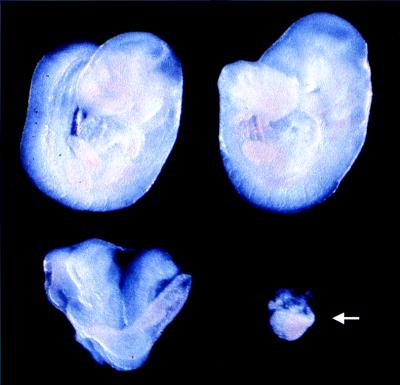
Whole mount preparations of E10.5 littermates from an intercross of RHA heterozygotes. Although normal embryos differ in the degree of differentiation, they have already advanced to organogenesis. In contrast, the mutant embryo appears as a small structureless mass of tissue (arrow). Genotypes of embryos, analyzed by Southern blot, are presented in Fig. 1E.
In Situ Hybridization.
The C-terminal Brachyury cDNA (nucleotides 1048–1764) (16) was amplified by PCR with 5′ primer (5′-CTGCAGTCCCATGATAACTGGTCTAGC-3′) and 3′ primer (5′-CCAGGATTTCAAAGTCACATATATGTTGTAG-3′) from a mouse embryonic cDNA library (CLONTECH). The 717-bp PCR product was cloned into a pCRII vector (Invitrogen) and the resulting plasmid pCT-1 was digested with XbaI or SpeI and transcribed in vitro with SP6 or T7 RNA polymerase (Stratagene) in the presence of labeled ribonucleoside triphosphates to generate either antisense or sense RNA probes, following the procedure recommended by the manufacturer. The plasmid, harboring HNF-3β cDNA (kindly supplied by James Darnell, Rockefeller University, New York), was digested with ClaI or NotI and transcribed with either T3 or T7 polymerase for either antisense or sense riboprobes as described above. In situ hybridization on 4-μm paraffin sections of embryos was performed essentially as described (16, 19). Embryo sections hybridized with sense probes for HNF-3β and Brachyury showed no signals above background levels.
Terminal Deoxynucleotidyltransferase-Mediated UTP End Labeling (TUNEL) Reaction.
Embryo sections were subjected to TUNEL reactions as described previously (15) with minor modifications. In brief, sections were deparaffinized in Histoclear (Fisher) and rehydrated by successive incubations in ethanol solutions (100%, 95%, and 70%, respectively) and diethyl pyrocarbonate-treated water. Embryo sections were then treated for 10 min at room temperature with proteinase K (20 μg/ml) in PBS containing 1% Triton X-100, and bleached in 0.1% H2O2 for 15 min at room temperature to destroy endogenous peroxidase activity. Nicks present in the DNA of apoptotic cells were end-labeled for 1 hr at 37°C with terminal deoxynucleotidyltransferase (40 units) (Boehringer Mannheim) and biotin 16-dUTP (5 μM) in a mixture (100 μl per slide) containing 30 mM Tris⋅HCl (pH 7.2), 140 mM sodium cacodylate, and 1 mM cobalt chloride. After blocking with 2% BSA in PBS, the slides were treated with ABC Vectastain reagent (Vector) for 30 min at room temperature, rinsed with 0.5% Triton X-100 in PBS, and then incubated for 15 min in a solution containing 0.3 mg/ml diaminobenzidine (Sigma) in 0.012% H2O2/PBS. After counterstaining with Gill’s hematoxylin for 2–4 sec, the slides were mounted with Permount (Fisher) and examined.
RESULTS
RHA Is Essential for Early Embryonic Development in Mice.
The RHA locus in mice was mutated by using ES cell technology (Fig. 1). A positive/negative selection strategy was used to disrupt exon II of the RHA gene, which contains the translation start codon ATG (6) (Fig. 1A). A targeting vector, containing the Neo gene as the positive selection marker and the herpes simplex virus tk gene as the negative selection marker (Fig. 1B), was electroporated into CJ7 ES cells (129/Sv), and clones carrying one mutant RHA allele (RHA−) were identified by Southern blot analysis. As depicted in Fig. 1C, XbaI digestion followed by Southern analyses of genomic DNAs from various ES cell lines are expected to yield signals 7.3 kb and 8.4 kb in length, corresponding to the RHA wild-type and mutant allele, respectively. As shown in Fig. 1D, among seven different G418/ganciclovir-resistant ES clones analyzed, only one ES clone (lane 6) exhibited equally intense Southern signals derived from RHA wild-type and mutant alleles of the predicted lengths, indicating that the mutation was introduced into one RHA allele by homologous recombination. All other signals were most likely derived from G418/ganciclovir-resistant clones that arose through the random integration of a single copy (lanes 1–5) or two copies (lane 7) of the targeting vector. From a total of 12 different RHA+/− ES cell lines isolated, two independent RHA+/− ES cell lines were injected into blastocysts (C57BL/6) and of the four chimeric mice born, three transmitted the RHA− allele to their progeny. Among mice examined up to 2 years of age, no abnormalities attributable to the heterozygous genotype have been observed.
Figure 1.

Targeted disruption of the RHA gene. (A) Restriction map of the 16.1-kb 5′ genomic region of the RHA gene (6). (A and B) □ represent the genomic regions used for genotyping by PCR and Southern blot analyses, respectively. Exons (I to V) are indicated by ■. Italic letters X and K represent restriction enzymes XbaI and KpnI. (B) Replacement vector. The arrows above Neo and tk indicate the direction of transcription of each selection marker. (C) Structure of mutant RHA gene allele resulting from homologous recombination. (D) Southern blot analysis of targeted ES cell lines. Among 7 clones represented, only one ES clone (lane 6) exhibited an equal intensity of Southern signals between wild-type and mutant RHA alleles of the predicted sizes (see below). (E) Genotypes of E10.5 embryos. In D and E, probe B (see A) was used to detect restriction fragments indicative of the predicted gene replacement event at the RHA locus. XbaI digestion should produce 7.3-kb and 8.4-kb genomic fragments for wild-type (wt) and mutant RHA alleles, respectively. (F) Genotypes of E7.5 embryos obtained by PCR analysis. The insertion of the Neo gene in the mutant RHA allele leads to an increase of 1.1 kb in size of the PCR product compared with that of the wild-type allele (180 bp).
In 20 litters of mice derived from RHA heterozygous intercross matings, no homozygous animals were identified, suggesting that homozygosity for the RHA mutant allele leads to embryonic lethality (Table 1). To determine the time and cause of embryonic lethality, embryos derived from intercrosses of heterozygotes of hybrid background (C57BL/6 × 129/Sv) were dissected at different stages of gestation. The genotypes were confirmed by Southern blot analysis with E9.5–19 embryos (Fig. 1E) or by the use of PCR assays with E6.5–8.5 embryos (Fig. 1F), as summarized in Table 1. E10.5 was the latest gestational age when the mutants could still be observed. At this stage, they were small embryonic masses exhibiting marked resorption (arrow in Fig. 2). Similar abnormalities were observed with all E9.5 null embryos (data not shown), suggesting an earlier onset of the mutant phenotype due to the loss of the RHA gene.
Table 1.
Genotype of embryos obtained from heterozygons mating
| Age | Number of genotype
|
|
|---|---|---|
| +/+ or +/− | −/− | |
| Neonate | 100 | 0 |
| E16.5 | 30 | 0 |
| E10.5 | 47 | 10 |
| E9.5 | 30 | 6 |
| E8.5 | 26 | 8 |
| E7.5 | 20 | 7 |
The genotype was determined either by Southern blot analysis with E9.5–E16.5 embryos and neonates or by PCR analysis with E7.5 or E8.5 embryos.
RHA Is Required for Normal Gastrulation.
To examine the effects of the RHA mutation in more detail, histological analyses after TUNEL reactions were performed with paraffin sections from E7.5 and E8.5 embryos. In addition to structural information, these analyses permitted the in situ localization of apoptotic cells (15). Most of the embryos used in these studies were from hybrid background crosses. The mutant phenotype was first observed at E7.5 (Fig. 3 B and C), a stage at which normal embryos exhibited elongated and characteristically shaped embryonic cups with distinct anterior/posterior and dorsal/ventral polarity (Fig. 3A). In a normal E7.5 embryo, gastrulation is well advanced and a mesodermal layer surrounds the embryonic ectoderm. Often the amnion is closed, the allantois is just forming, and the extraembryonic ectoderm is lifted up into the two-layered chorion. The parietal and visceral endoderm form squamous sheets around the embryo proper. In contrast to this, mutant embryos were smaller in size and significantly delayed in development (Fig. 3 B and C). Morphologically they resembled embryos from E6.5 litters and contained a small two-layered egg cylinder. The parietal endodermal cells were rounded, and a cuboidal layer of visceral endoderm surrounded the entire conceptus. No mesodermal layer could be distinguished in sections and no amnion was present. However, the extraembryonic ectoderm formed a distinct ectoplacental cone and, in some embryos, a thickening of the embryonic ectoderm was observed (arrow in Fig. 3C). Although few dying cells were observed in the control embryos (arrows in Fig. 3A), numerous apoptotic bodies were present in the mutants, localized predominantly in the embryonic ectoderm. The apoptotic bodies were frequently clustered and also present in the proamniotic cavity (Fig. 3B).
Figure 3.
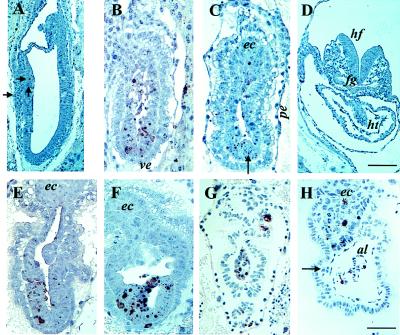
Histological analysis of E7.5 and E8.5 embryos. Paraffin sections (4 μm) of embryos were subjected to TUNEL reaction as described (15). Embryos in A and F were obtained from inbred strains. (A) E7.5 wild-type embryo. Solid arrows point to apoptotic bodies present in the anterior embryonic ectoderm. The section in A is adjacent to the sections presented in Fig. 4 A and C. (B and C) E7.5 mutant littermates. The section in C is adjacent to the sections presented in Fig. 4 B and D. The arrow points to a presumptive primitive streak. (D) E8.5 wild-type embryo. (E and F) E8.5 mutant embryos, littermates of the embryo shown in D. (G and H) E7.5 and E8.5 mutant embryos, respectively, from the outbred strain Black Swiss. The solid arrow in H points to a presumptive blood island. [Bar = 100 μm in D (also applies to A) and 50 μm in H (also applies to B, C, E, F, and G).] Abbreviation used in Figs. 3 and 4 are as follows: ab, allantoic bud; al, allantois; amn, amnion; ec, ectoplacental cone; ee, embryonic ectoderm; ems, embryonic mesoderm; fg, foregut; hf, headfold; ht, heart; nch, notochordal plate; pe, parietal endoderm; ve, visceral endoderm.
More striking differences were noted between mutants and controls at E8.5. At this stage, normal littermates have already initiated organogenesis (Fig. 3D), whereas RHA mutants were developmentally similar to the E7.5 mutants, manifesting only a slight increase in size (Fig. 3 E and F). Although the embryonic portion of the visceral endoderm appeared flattened and the embryonic ectoderm acquired a pseudostratified epithelial morphology, no typical mesoderm was detected. Apoptotic cell death was profound in cells constituting the embryonic ectoderm.
To explore the possible contribution of genetic background to the observed effects of the RHA mutation, the mutant RHA allele was backcrossed into the outbred strain Black Swiss. Unexpectedly, the observed phenotype became more severe. At E7.5, the embryonic ectoderm was almost entirely absent and only a few dying cells remained (Fig. 3G). By E8.5, only the extraembryonic portion remained and exhibited some degree of differentiation (Fig. 3H). Independent of the absence of morphologically discernible embryonic mesoderm, extraembryonic mesoderm was generated and gave rise to allantois, the lining of the exocoelom, and even to a few small yolk sac blood island-like structures. However, the ectoplacental cone remained relatively undifferentiated and apoptotic cells were abundant. On the basis of these observations, we conclude that the deletion of the RHA gene affects the activity of genes involved specifically in the survival of the embryonic ectoderm during the early stage of gastrulation, and more importantly, in its ability to undergo proper differentiation.
Induction of Gastrulation Takes Place in RHA-Deficient Embryos.
Although no embryonic mesodermal layer was observed by histological analysis of RHA-deficient embryos, extraembryonic mesoderm was present and differentiated into allantois, the mesodermal layer of the yolk sac, and blood island-like structures. These observations indicate that the early onset of gastrulation, although delayed, is not affected by the loss of RHA function. The induction of gastrulation in RHA mutants was confirmed by RNA in situ hybridization for the expression of Brachyury (T gene) and HNF-3β, two molecular markers specific for early stages of gastrulation (16–22). As expected from the morphology of the embryos, both genes were expressed in the mutant embryos as early as E7.5 and more strongly at E8.5.
At E7.5, mutant embryos expressed both Brachyury and HNF-3β in a pattern consistent with the early onset of gastrulation. A small group of primitive embryonic ectodermal cells expressed Brachyury (Fig. 4D), and weak HNF-3β expression was observed in the same portion of the adjacent embryo section (Fig. 4B). Most likely this expression is indicative of the differentiation of an early primitive streak and the initiation of an anterior/posterior axis. In addition, the entire visceral endoderm expressed HNF-3β, a property characteristic of normal pre- and early streak stages of gastrulation. In contrast to the RHA mutants, the expression of both genes in normal littermates reflected the advanced progression of gastrulation, including the formation of the node and notochordal plate (Fig. 4 A and C). At E8.5, in situ hybridization analysis of mutant embryos confirmed the lack of progression of gastrulation and differentiation. The patch of ectoderm cells expressing Brachyury (Fig. 4H) was just slightly larger than that found in E7.5 mutants (Fig. 4D). The expression of HNF-3β was also somewhat enhanced, notably in cells localized anterior to the ectodermal cells that expressed Brachyury (compare Fig. 4 F and H). This is reminiscent of the normal expression pattern of HNF-3β in the anterior part of the primitive streak (19–22). However, there was no delamination of cells out of the aberrant primitive streak and no embryonic mesodermal layer could be detected. From these observations, we conclude that RHA affects the activity of genes critical for the differentiation of embryonic ectoderm and hence the normal progression of gastrulation.
Figure 4.
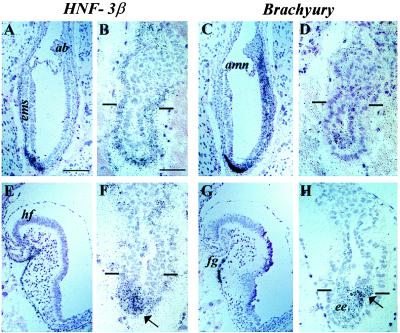
Expression of HNF-3β and Brachyury in E7.5 (A–D) and E8.5 (E–H) embryos. Adjacent sections of wild-type embryos (A, C, E, and G) and their mutant littermates (B, D, F, and H) were hybridized with HNF-3β (A, B, E, and F) and Brachyury (C, D, G, and H) antisense probes as described (16, 19). For all images, anterior is to the left, and posterior is to the right. Arrows in the mutants point to aberrant primitive streak, and horizontal lines show the presumptive embryonic–extraembryonic border. [Bar = 100 μm in A (also applies to C, E, and G) and 50 μm in B (also applies to D, F, and H).]
DISCUSSION
This study demonstrates that RHA is essential for embryonic development in mice. It establishes a developmental role for RHA in mammals. Depletion of RHA resulted in a mutant phenotype as early as E7.5, and the embryos were completely resorbed by E10.5. The early embryonic lethality was associated with the inability of the embryonic ectoderm to differentiate normally during gastrulation. This observation suggests that RHA and its Drosophila homologue MLE play important roles in development.
In Drosophila, MLE plays critical role(s) in the development of male flies (3). However, at the post-developmental stage, as-yet-unidentified sets of factor(s) present in both sexes may recruit the function of MLE to increase the expression of certain genes including para, an X-linked gene coding for a sodium channel (23–28). The higher genetic complexity of mammals makes it possible to envision more diverse role(s) for RHA than MLE in transcriptional regulation processes. The phenotypes observed in RHA mutant mouse embryos may indicate the earliest developmental stage when RHA begins to act in conjunction with its interacting transcription factor(s), functionally resembling the male-specific lethal proteins of Drosophila (24–28). It is presently unknown which transcriptional activation processes require the function of RHA during development and differentiation. The cAMP signaling pathway is currently the only demonstrated process that requires RHA to provoke target gene activation (7).
There are a number of factors participating in the cAMP signaling pathway, which include cAMP-dependent protein kinase (PKA), cAMP-responsive element binding protein (CREB), cAMP-responsive element modulating protein (CREM), CBP, and RHA. If RHA specifically participates in this pathway during development, the deletion of any one of the genes described above should produce a phenotypic alteration similar to that observed in the RHA mutant. Alternatively, the cAMP signaling pathway may be one among a number of other pathways that require the function of RHA. If this were the case, the deletion of genes involved in the cAMP signaling pathway could yield a milder phenotype relative to the deletion of the RHA gene.
Previous studies have shown that mice containing targeted mutations in genes encoding cAMP-responsive element binding protein and the regulatory subunit II of cAMP-dependent protein kinase are viable (29, 30). Interestingly, these mutations lead to the increased expression of cAMP-responsive element modulating protein and the regulatory subunit I of cAMP-dependent protein kinase in such animals. These compensatory levels of expression may be responsible for the viability of such mutant mice. An important role of CBP in development has been suggested by the correlation of the Rubinstein–Taybi syndrome with the loss of one functional copy of the CBP gene (31). Rubinstein–Taybi syndrome patients are characterized by abnormal pattern formation such as facial abnormalities, broad thumbs and big toes, and mental retardation (32). In Drosophila, CBP functions as a coactivator of cubitus interruptus (ci), an important gene involved in normal pattern formation during embryogenesis (33). Possibly, mutant mice devoid of the CBP gene or those that are heterozygous for CBP or heterozygous for both the CBP and RHA genes may provide clues to the biological role of CBP and its functional interaction with RHA during development.
Acknowledgments
We are indebted to Karen Witty-Blease and Scott Kerns of the Molecular Cytology Core Facility of Memorial Sloan–Kettering Cancer Center for their technical help. This research was supported in part by a grant from the New York Heart Association to J.H. and in part by the Memorial Sloan–Kettering Cancer Center Support Grant (CA-08748).
ABBREVIATIONS
- RHA
RNA helicase A
- MLE
Drosophila maleless protein
- CBP
cAMP-responsive element binding protein (CREB)-binding protein
- TUNEL
terminal deoxynucleotidyltransferase-mediated UTP end labeling
- ES cells
embryonic stem cells
- En
embryonic day n
References
- 1.Lee C G, Chang K A, Kuroda M I, Hurwitz J. EMBO J. 1997;16:2671–2681. doi: 10.1093/emboj/16.10.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee C G, Hurwitz J. J Biol Chem. 1992;267:4398–4407. [PubMed] [Google Scholar]
- 3.Lee C G, Hurwitz J. J Biol Chem. 1993;268:16822–16830. [PubMed] [Google Scholar]
- 4.Zhang S, Grosse F. Biochemistry. 1994;33:3906–3912. doi: 10.1021/bi00179a016. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S, Macke H, Grosse F. J Biol Chem. 1995;270:16422–16427. doi: 10.1074/jbc.270.27.16422. [DOI] [PubMed] [Google Scholar]
- 6.Lee C G, Eki T, Okumura K, Soares V C, Hurwitz J. Genomics. 1998;47:365–371. doi: 10.1006/geno.1997.5139. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima T, Uchida C, Anderson S F, Lee C G, Hurwitz J, Parvin J D, Montminy M. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 8.Kelley R L, Kuroda M I. Science. 1995;270:1607–1610. doi: 10.1126/science.270.5242.1607. [DOI] [PubMed] [Google Scholar]
- 9.Lyon M F. Nature (London) 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda M I, Meller V H. Cell. 1997;91:9–11. doi: 10.1016/s0092-8674(01)80003-9. [DOI] [PubMed] [Google Scholar]
- 11.Solter D, Wei G. Genes Dev. 1997;11:153–155. doi: 10.1101/gad.11.2.153. [DOI] [PubMed] [Google Scholar]
- 12.Lyon M F. Nature (London) 1996;379:116–117. doi: 10.1038/379116a0. [DOI] [PubMed] [Google Scholar]
- 13.Lee K A. Curr Opin Cell Biol. 1991;3:953–959. doi: 10.1016/0955-0674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- 14.Montminy M. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sason S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrman B G, Labeit S, Poustka A, King T R, Lehrach H. Nature (London) 1990;343:617–622. doi: 10.1038/343617a0. [DOI] [PubMed] [Google Scholar]
- 17.Wilkinson D G, Bhatt S, Herrmann B G. Nature (London) 1990;343:657–659. doi: 10.1038/343657a0. [DOI] [PubMed] [Google Scholar]
- 18.Kispert A, Hermann B G. Dev Biol. 1994;161:179–193. doi: 10.1006/dbio.1994.1019. [DOI] [PubMed] [Google Scholar]
- 19.Monaghan A P, Kaestner K H, Grau E, Schutz G. Development (Cambridge, UK) 1993;119:567–578. doi: 10.1242/dev.119.3.567. [DOI] [PubMed] [Google Scholar]
- 20.Lai E, Prezioso V R, Tao W, Chen W S, Darnell J E. Genes Dev. 1991;5:416–427. doi: 10.1101/gad.5.3.416. [DOI] [PubMed] [Google Scholar]
- 21.Ruiz I, Altaba A, Prezioso V R, Darnell J E, Jessel T M. Mech Dev. 1993;44:91–108. doi: 10.1016/0925-4773(93)90060-b. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki H, Hogan B L M. Development (Cambridge, UK) 1993;118:47–59. doi: 10.1242/dev.118.1.47. [DOI] [PubMed] [Google Scholar]
- 23.Kernan M J, Kuroda M I, Kreber R, Baker B S, Ganetzky B. Cell. 1991;66:949–959. doi: 10.1016/0092-8674(91)90440-a. [DOI] [PubMed] [Google Scholar]
- 24.Palmer M J, Mergner V A, Richman R, Manning J E, Kuroda M I, Lucchesi J C. Genetics. 1993;134:545–557. doi: 10.1093/genetics/134.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bashaw G J, Baker B S. Development (Cambridge, UK) 1995;121:3245–3258. doi: 10.1242/dev.121.10.3245. [DOI] [PubMed] [Google Scholar]
- 26.Kelley R L, Solovyeva I, Lyman L M, Richman R, Solovyev V, Kuroda M I. Cell. 1995;81:1–20. doi: 10.1016/0092-8674(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 27.Zhou S, Yang Y, Scott M J, Pannuti A, Fehr K C, Eisen A, Koonin E V, Fouts D L, Wrightsman R, Manning J E, Lucchesi J C. EMBO J. 1995;14:2884–2895. doi: 10.1002/j.1460-2075.1995.tb07288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorman M, Franke A, Baker B S. Development (Cambridge, UK) 1995;121:463–475. doi: 10.1242/dev.121.2.463. [DOI] [PubMed] [Google Scholar]
- 29.Hummler E, Cole T J, Blendy J A, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. Proc Natl Acad Sci USA. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummings D E, Brandon E P, Planas J V, Motamed K, Idzerda R L, McKnight G S. Nature (London) 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 31.Petrij F, Giles R H, Dauwerse H G, Saris J J, Hennekam R C, Masuno M, Tommerup N, van Ommen G J, Goodman R H, Peters D J, et al. Nature (London) 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein J H, Taybi H. Am J Dis Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- 33.Akimura H, Chen Y, Hou D X, Nonaka M, Smolik S M, Armstrong S, Goodman R H, Ishii H. Nature (London) 1997;386:735–738. doi: 10.1038/386735a0. [DOI] [PubMed] [Google Scholar]
- 34.Joyner A L. Gene Targeting. Oxford: IRL; 1993. [Google Scholar]