

Abstract
Insulin-dependent diabetes mellitus is an autoimmune disease, under polygenic control, manifested only when >90% of the insulin-producing β cells are destroyed. Although the disease is T cell mediated, the demise of the β cell results from a number of different insults from the immune system. It has been proposed that foremost amongst these effector mechanisms is CD95 ligand-induced β cell death. Using the nonobese diabetic lpr mouse as a model system, we have found, to the contrary, that CD95 plays only a minor role in the death of β cells. Islet grafts from nonobese diabetic mice that carry the lpr mutation and therefore lack CD95 were protected only marginally from immune attack when grafted into diabetic mice. An explanation to reconcile these differing results is provided.
The nonobese diabetic (NOD) mouse model of spontaneous diabetes has provided an important tool to study the initiation and progression of diabetes, which are almost impossible to follow in humans (1–3). The current consensus defines a three-step process in diabetes development in NOD mice. First, CD8+ T cells are essential to initiate islet infiltration (3, 4) and are one of the earliest cell types to be seen in the islets (5). Second, CD4+ T cells and CD8+ T cells infiltrate and surround the islets, which remain relatively intact. Third, all of the β cells eventually are killed, and diabetes occurs. Although CD4+ T cells are directly pathogenic in the NOD mouse model (3, 6, 7), it is clear that CD8+ T cells are important in disease acceleration (8–10).
The way in which T cells kill islets is complex, and diverse mechanisms probably contribute to the demise of the β cell. Injury may result from CD4+ or CD8+ T cells secreting cytokines in response to MHC-restricted recognition of β cell antigens (11). Cytokines such as interleukin 1β can induce damaging free radicals like nitric oxide (12) or can activate the function of cell death molecules (CD95 and tumor necrosis factor receptor I) on the islets, allowing the β cell to play a part in its own destruction (13). Studies with perforin-deficient NOD mice indicated that cytotoxic, CD8+ T cell attack is crucial in the final progression to diabetes (10). All of these effectors (perforin, nitric oxide, CD95 ligand, and tumor necrosis factor) are able to set in motion a pathway leading to apoptosis (13), and several studies have shown that this process is also a characteristic of β cells (14–17).
Recently, the idea that β cell death results primarily from CD95 ligand (CD95L)-induced apoptosis has received considerable support. CD95 is expressed constitutively in most tissues and is up-regulated at sites of inflammation (18). Pancreatic islet cells are no exception, and, although normal islets do not express CD95, both human (19) and mouse (20) islet cells can synthesize the receptor in vitro, after incubation with interleukin 1β, or in vivo, after leukocyte infiltration (21, 22). The induced islets were sensitive to apoptotic cell death mediated by agonistic anti-CD95 antibodies (19, 20). Two groups used a CD95-deficient variant (lpr) of the NOD mouse and showed that diabetogenic NOD spleen cells (23) or a NOD-derived CD8+ T cell clone specific for β cells (22) did not cause diabetes when transferred into NODlpr recipients. Separately, they reached the conclusion that induction of CD95 expression on β cells, and its engagement by CD95L on infiltrating T cells, represented the main pathogenic mechanism in autoimmune diabetes.
The lpr mutation in mice results in lymphadenopathy, the intensity of which is determined by genetic background (24). NODlpr mice had huge numbers of the CD4−CD8−B220+ α/β T cells characteristic of animals carrying the lpr mutation (23). Using this mutation, we also have studied the role of CD95 as an effector of β cell death in NOD mice. We were concerned that the lymphadenopathy of NODlpr animals might influence our results, so we took an approach that differed from that of others (22, 23) and came to a different conclusion. CD95-mediated death of β cells, in our experiments, proved to be only a minor effector in the development of diabetes.
MATERIALS AND METHODS
Mice.
NOD/Lt and C3H/Hejlpr mice were from our special pathogen free facility at Kew (Victoria, Australia). Our mouse rooms are free from most viral pathogens except rotavirus. The lpr mutation from the C3H/Hejlpr strain was backcrossed to the NOD/Lt background for seven generations, at which time 7 of 13 (53%) females became diabetic by 280 days. Brother × sister matings were done to generate NOD/Ltlpr (NODlpr) mice. The presence of the lpr mutation at each backcross was detected by PCR analysis as described (25). Although the NODlpr mice were derived from a seventh generation backcross, there was no problem with them making an allo-reponse to NOD antigens because NOD islets remained healthy and uninfiltrated in NODlpr recipients even 64 days after grafting.
Fetal Pancreas Grafts.
Pancreata of 15- to 18-day-old donor embryos were cultured on 0.45-μm Millipore membranes (type HV) in DMEM plus 15% fetal calf serum (FCS) for 10–14 days and were grafted under the kidney capsule of NOD or NODlpr mice. Although the H-Y response in NOD mice is weak (26), we typed the grafts for sex, but, because no difference was seen in the response of female recipients to male or female grafts, the results from these experiments were pooled. The NOD and NODlpr fetal pancreas cultures were analyzed by histology before grafting, and no obvious difference in their appearance or insulin expression was seen.
Transfer of Diabetes and Measurement of Diabetes Incidence.
Transfer of diabetes to irradiated (850R) NOD and NODlpr mice was done by injecting 20 million splenocytes from diabetic NOD/Lt female donors in 0.3 ml of PBS into the tail vein (27). FCS was excluded from the inoculum if animals had been grafted previously with pancreas cultures grown in DMEM/FCS, to prevent anaphylaxis on reexposure to FCS. Urine glucose readings were done at daily intervals beginning 2 weeks after transfer. Three consecutive readings of 60–110 mM were taken to indicate the onset of diabetes. If diabetic mice were kept, they were maintained on insulin (1 unit of isophane insulin, injected s.c. twice a day).
CSFE-Labeling of Splenocytes for Transfer to Mice.
Preparation and labeling of splenocytes with 5,6-carboxy-succinimidyl-fluorescein-ester (CFSE) (Molecular Probes, Eugene, Oregon) was done as described (28). In brief, donor spleen cells were washed in HEM/FCS (10%) and were treated with anti-heat-stable antigen antibody (J11D) for 30 min at 4°C followed by complement treatment to remove red cells. Cells were washed twice in PBS containing 0.1% BSA, were resuspended at 107 cells/ml, and were filtered through 100-μm nylon mesh. CFSE (5 mM in DMSO) was added at 2 μl per 107 cells/ml, and cells were incubated in the dark for 10 min at 37°C, after which they were washed twice in HEM/FCS (10%) and were resuspended at 0.5 × 107 cells/ml in PBS. Immediately before injection into the tail vein of mice, the cells were passed through a 40-μm nylon mesh to remove clumps. Recipient mice were aged between 5 and 7 weeks. At this age, the NODlpr animals had started to accumulate the CD4−CD8−B220+ α/β T cells characteristic of lpr mice but not to such an extent that cell numbers in the spleen were grossly disproportionate to those in normal NOD mice.
Flow Cytometry.
Analysis of CFSE-labeled cells was as follows. One or four days after transfer of CFSE labeled cells, mice were killed by asphyxiation in CO2. Spleens were chopped and pushed through a sieve in 10 ml of HEM/FCS (10%). Leukocyte counts were done immediately on a phase contrast microscope to distinguish red cells. This gave the total spleen leukocyte yield. Cells were prepared for FACS analysis and were stained with propidium iodide at 0.5 μg/ml, and the percent of live spleen cells positive for CFSE was determined in a FACScan analyzer (Becton Dickinson) using lysis ii software (Becton Dickinson). Cells also were stained for T cell markers by using phycoerythrin conjugates of the antibodies GK1.5 (anti-CD4), YTS 169.4 (anti-CD8) (Caltag, South San Francisco, CA), and RA3–6B2(anti-B220) (PharMingen).
Histology and Immunohistochemistry.
Procedures for histology and immunohistochemistry on Bouin’s solution fixed paraffin sections have been detailed (29). Guinea pig antiinsulin and rabbit anti-somatostatin or anti-glucagon antibodies were from Dako. The secondary antibodies, goat anti-guinea pig (from Chemicon) and sheep anti-rabbit (Dako), were horseradish peroxidase conjugates, and color development was with diaminobenzidine.
RESULTS
NODlpr Mice.
The mice in our NODlpr colony were similar to those described by others (22, 23). They did not become diabetic by 150 days of age, and at this time they had healthy pancreatic islets with only occasional infiltrating cells. Infiltrating leukocytes were seen in other organs such as lung, kidney, salivary gland, or liver, and the animals suffered from a dramatic lymphadenopathy that was apparent as early as 5 weeks of age. By 6 months, the animals were unwell and therefore were not kept beyond this age.
Transfer of Diabetogenic Cells to NODlpr Mice Does Not Cause Diabetes.
The fact that NODlpr mice did not develop diabetes or insulitis most likely was related to their perturbed immune system resulting from the lpr mutation. Because our interest was in the relevance of CD95 to β cell death, we used the diabetes transfer system (27) to test the resistance of NODlpr islets to autoimmune destruction. Irradiated NOD mice and NODlpr mice were used as recipients of spleen cells from recently diabetic NOD mice. All of the NOD recipients (7/7) became diabetic within 16–23 days as expected, but none of the NODlpr mice (0/11) did so, not even by 53 days. Histology of the NODlpr pancreas showed healthy islets, although some did have a few infiltrating lymphocytes (not shown). These results are the same as those reported by Itoh et al. (23).
Transfer of Diabetogenic Cells to NODlpr Mice that Carry NOD Islet Grafts.
It was surprising that transfer of diabetogenic cells to NODlpr mice resulted in little or no pathology in the islets. It was expected that such cells would home to the islets but would be unable to kill the β cells that lacked CD95. We therefore grafted young (6–10 week) NODlpr male mice with NOD or NODlpr fetal pancreas grafts at either pole of the kidney. The grafts were left for 2 weeks, by which time they had formed healthy tissue with many islets and little infiltration because the islets of young NOD males are not subject to much pathology. The mice were irradiated (850R) and given diabetogenic spleen cells in the usual way. If CD95 deficiency on the islets was responsible for the lack of killing of β cells by the diabetogenic cells, then the CD95-sufficient NOD islet grafts should be damaged but the NODlpr islet grafts and NODlpr pancreas islets should not. The mice, however, did not develop diabetes, and healthy grafts with many islet clusters were found at the graft sites (Table 1, Experiment 1 and Fig. 1A and B). Histological analysis showed that NOD islet grafts looked as healthy as NODlpr islet grafts or the NODlpr pancreas, and no infiltrating cells were seen (Fig. 1 C and D). A second experiment using young (4 week) female NODlpr recipients gave the same result (Table 1, Experiment 2). This indicated that there was a problem in the experimental design used to induce diabetes by transferring diabetogenic spleen cells to NODlpr recipients.
Table 1.
Transfer of diabetogenic spleen cells to NOD and NODlpr mice grafted with fetal pancreas
| Donor cells (female) | Recipient mice | Manipulation* | No. diabetic | Day after receiving transferred cells |
|---|---|---|---|---|
| Experiment 1 | ||||
| NOD diabetic spleen cells | NODlpr males | Islet grafted | 0/7 | 56, 56, 56, 64, 64, 64, and 64 |
| NOD nondiabetic spleen cells | NODlpr males | Islet grafted | 0/4 | 56, 56, 56, and 56 |
| NOD diabetic spleen cells | NOD males | Not grafted | 3/3 | 20, 20, and 68 |
| Experiment 2 | ||||
| NOD diabetic spleen cells | NODlpr females | Islet grafted | 0/3 | 54, 54, and 54 |
| NOD diabetic spleen cells | NOD females | Not grafted | 3/3 | 17, 17, and 20 |
| Experiment 3 | ||||
| NOD diabetic spleen cells | NOD males | Islet grafted | 7/9 | 15, 17, 18, 20, 21, 23, and 34 |
| NOD nondiabetic spleen cells | NOD males | Islet grafted | 0/5 | 56, 56, 59, 59, and 61 |
Mice were grafted with NOD and NODlpr fetal pancreas islet grafts at either pole of the kidney capsule and rested for 2–3 weeks. They then were irradiated (850R) and given 2 × 107 spleen cells i.v. Control NOD mice were not grafted.
Figure 1.

Integrity of NOD and NODlpr fetal pancreas islet grafts in NODlpr mice. NOD (A and C) or NOD lpr (B and D) islets were grafted into NODlpr recipients, which subsequently were given diabetogenic spleen cells. (A and B) Appearance of islets under the kidney capsule 56 days after transfer of cells. (C and D) Insulin staining of tissue sections from the grafts. (A and B, ×10; C and D, ×100.)
Inefficient Survival of NOD Spleen Cells Injected into NODlpr Mice.
We were concerned that the NOD diabetogenic spleen cells that were transferred to NODlpr recipients were in some way being deleted, diluted, or removed before they could express their diabetogenic potential. To test this, spleen cells from NOD mice were labeled with the fluorescent dye CFSE and were transferred to young, irradiated recipient NOD or NODlpr mice. The transferred splenocytes underwent dramatic proliferation, and it appeared that NODlpr recipients had fewer CFSE-labeled cells than NOD recipients, with CD4+ T cells being preferentially depleted. Because the splenocytes were proliferating in the irradiated hosts, it was not possible to measure accurately the number of cells that had been removed. We therefore transferred cells from nondiabetic NOD mice into nonirradiated NOD or NODlpr recipients to avoid the problem of donor cell proliferation associated with irradiation. After 24 hours, CFSE-labeled NOD cells were present in the spleens of both NOD and NODlpr recipients (Table 2). There were marginally less CFSE+ cells in NODlpr recipients compared with NOD recipients. On day 4, the NOD recipients had the same number of cells as at day 1, but NODlpr mice had lost even more cells, so they now had 80% less than NOD recipients. Analysis of the subsets of surviving CFSE-labeled lymphocytes showed that CD4+ T cells were particularly prone to depletion, and 90% less of this subset was present in NODlpr recipients. On the other hand, NODlpr splenocytes were not depleted preferentially when transferred to NODlpr hosts (Table 2). Apparently, NOD spleen cells were unable to survive in NODlpr mice. These results implied that the number of NOD diabetogenic spleen cells normally used to transfer diabetes to NOD recipients (2 × 107 in this study) might be insufficient to transfer diabetes to NODlpr recipients because so many would be lost before being able to attack the islets.
Table 2.
Inefficient survival of NOD splenocytes injected into NODlpr recipients
| Transfer protocol, donor to recipient* | Total no. of CFSE+ cells in spleen | No. of CFSE+ CD4+ T cells in spleen | No. of CFSE+ CD8+ T cells in spleen | No. of CFSE+ B220+ B cells in spleen |
|---|---|---|---|---|
| At day 1 | × 106 | × 104 | × 104 | × 104 |
| NOD to NOD | 1.0 (1–1.2) | |||
| NOD to lpr | 0.7 (0.5–0.9) | |||
| lpr to lpr | 1.5 (1.3–1.9) | |||
| At day 4 | ||||
| NOD to NOD | 1.0 (0.9–1.3) | 15 (12–16) | 6 (5–7) | 52 (25–80) |
| NOD to lpr | 0.2 (0.2) | 1.6 (1.5–1.8) | 1 (1) | 12 (11–15) |
| lpr to lpr | 1.2 (1.1–1.4) |
Mice were injected with 2 × 107 CFSE-labeled splenocytes from nondiabetic NOD or NODlpr donors. Results represent the average from three mice, with the range given in parentheses.
Transfer of Diabetogenic Cells to NOD Mice that Carry NODlpr Islet Grafts.
The transfer of diabetogenic spleen cells into NODlpr recipients was hampered by removal of the incoming cells in the NODlpr host. To elucidate the importance of CD95 in the mechanism of β cell death, it was necessary to graft NODlpr fetal pancreas islets into NOD mice and transfer diabetogenic cells into an environment in which they would not be killed. We therefore grafted young NOD male mice with NOD or NODlpr fetal pancreas grafts at either pole of the kidney as described above, waited 2 weeks, and then, after irradiating these mice, transferred in NOD-diabetogenic spleen cells. In this case, seven of nine mice became diabetic by day 15–34 (Table 1, Experiment 3). Grafts were recovered and assessed by their appearance under the kidney capsule and by histological examination. The NODlpr graft sites, under the kidney capsule, were very small but more distinct than the NOD grafts, which looked more like a scar than a graft. Histological examination showed that both NOD and NODlpr grafts had undergone immune attack and were invaded by infiltrating lymphocytes (Fig. 2A and D or B and E). The NODlpr grafts had slightly more β cell mass than the NOD grafts, but this was disorganized and the β cells had variable, faint insulin staining. So compared with NOD grafts, the NODlpr grafts seemed to fare better under immune attack. Control grafts, recovered from mice that had received spleen cells from nondiabetic donors, had many large, healthy islets under the kidney capsule with intact organization, good insulin staining, and minimal infiltration (Fig. 2 C and F).
Figure 2.
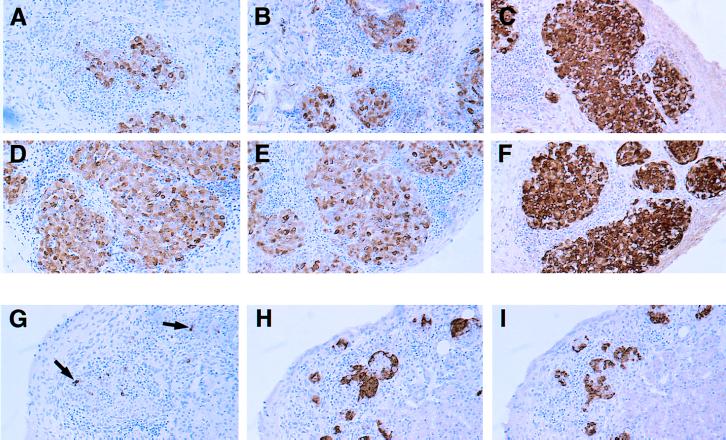
Histopathology of NOD or NODlpr islet grafts in diabetic NOD mice. Two examples of NOD (A and B) and NODlpr (D and E) islet grafts are shown at 21 or 15 days after transfer of diabetogenic spleen cells. The grafts from NOD mice that received nondiabetic spleen cells also are shown: NOD (C) and NODlpr (F) 61 days after transfer. (G–I) Appearance of NODlpr fetal pancreas islet grafts 30 days after being grafted under the kidney capsule of spontaneously diabetic female recipients. Sections were analyzed by immunohistochemistry (brown color) for insulin (A–G), somatostatin (H), and glucagon (I) and were counter-stained with haematoxylin. Arrows in G indicate occasional remaining β cells. (×100.)
NODlpr Islet Grafts Cannot Cure Spontaneously Diabetic Female NOD Mice.
Although the NODlpr fetal pancreas grafts were damaged by autoimmunity, they did survive slightly better than NOD grafts. A stringent test to see whether absence of CD95 could protect β cells against immune attack was to examine whether NODlpr islet grafts could reverse diabetes in spontaneously diabetic mice. Recently, diabetic female NOD mice were grafted separately with NOD or NODlpr fetal pancreas grafts and were maintained on twice-daily insulin injections. One group was analyzed after 11 days, and another was analyzed after 30 days. At the 11-day time point, four of five NOD graft sites and six of six NODlpr graft sites were recovered. Both graft types were infiltrated heavily, but the NODlpr grafts appeared somewhat less damaged than NOD grafts (results not shown). The grafts looked similar to those in the aforementioned experiments in which diabetogenic cells were transferred into NOD mice carrying NOD and NODlpr islet grafts. At the 30-day time point, none of the mice had recovered from their diabetes. Eight of nine NODlpr grafts were recovered, but none of the two NOD grafts were found. The NODlpr graft sites, under the kidney capsule, were hazy in appearance and contained very small islets. Histological examination showed the presence of infiltrating leukocytes and very small islet structures. On staining with antibodies to the three main islet cell types (insulin, somatostatin, and glucagon-expressing cells), it was found that the small islets were comprised mainly of somatostatin and glucagon secreting cells (Fig. 2 H and I). Sometimes β cells were seen, but these expressed very little insulin (Fig. 2G, arrows). So, although NODlpr β cells could resist autoimmune attack somewhat better than NOD β cells, they could not reverse diabetes in spontaneously diabetic recipients and eventually were destroyed.
DISCUSSION
We have used the natural mutation lpr in the CD95 gene to study the importance of this death receptor in β cell destruction. Our initial findings confirmed those of others, in that NODlpr mice did not develop diabetes or insulitis, nor was it possible to transfer diabetes to NODlpr recipients (22, 23). At first impression, this seemed to implicate CD95 as the major effector in the death of β cells. In our experiments, islets of NODlpr mice that had received NOD diabetogenic cells were remarkably free of any infiltrating lymphocytes and looked healthy. This result was surprising because diabetogenic donor cells contain effector T cells, and the expected outcome was that these would home to the pancreas islets but could not kill β cells that lacked CD95. When NODlpr mice were grafted with normal NOD islets and were given diabetogenic spleen cells, the NOD islets (which expressed CD95) were unharmed, suggesting that some other factor was preventing the islets from being attacked. This result is not consistent with the conclusions of Itoh et al. (23), who proposed that CD95 was important in initiation of insulitis.
The lack of insulitis in the islets of NODlpr recipients given diabetogenic spleen cells made us suspect the validity of the transfer model in this system. We found that CFSE-labeled, NOD spleen cells were depleted preferentially in irradiated and unirradiated NODlpr hosts and CD4+ T cells particularly were targeted. The lymphoproliferative disorder of lpr mice is associated with massive up-regulation of CD95L on CD4−CD8−B220+ α/β T cell population and these cells can kill CD95-expressing cell lines in vitro (30, 31). This ligand might have mediated the deletion of naive and activated CD95+ NOD donor T cells in our experiments because in vitro studies showed that naive T cells express CD95, and this can act as a death molecule if crosslinked by CD95L (32). In vivo, lpr bone marrow cells can cause graft-versus-host disease when injected into syngeneic recipients (33, 34), and one study found that the CD95-mediated liver damage that occurred during this graft-versus-host disease was associated with CD8+ lpr T cells (35). Another in vitro study showed that lpr CD4+ T cells expressed higher levels of CD95L on restimulation with superantigen (36). So, all populations of lpr T cells appear capable of expressing abnormally high levels of CD95L. In our studies, the cytotoxic effects of CD95L from the lpr hosts could account for the depletion of the NOD donor splenocytes in both unirradiated and irradiated NODlpr hosts because we have detected small numbers of T cells (1–3 × 106 T cells per spleen) in both irradiated NOD and NODlpr mice (J.A., unpublished results). At any rate, whatever the mechanism, our studies with CFSE-labeled NOD splenocytes indicated that NODlpr hosts could reject NOD donor cells, which presumably included the activated, effector T cells needed to transfer diabetes.
Because of the problems associated with transferring diabetes to NODlpr recipients, we tested the survival of CD95-deficient grafts in diabetic NOD hosts. Using this experimental procedure, it was found that islets lacking CD95 were subject to autoimmune destruction in the same way as normal NOD islets. It appeared, however, that grafts lacking CD95 suffered damage more slowly, although eventually they were destroyed. The slight protection afforded by the absence of CD95 led us to conclude that this molecule plays only a minor role in the death of β cells during the effector phase of autoimmunity in the NOD mouse. Chervonsky et al. (22), on the other hand, made a case for CD95 being the main pathogenic molecule in autoimmune diabetes. They based this on experiments in which a CD8+ T cell clone, specific for β cells, was transferred to irradiated NOD or NODlpr recipients. These T cells infiltrated the islets of NODlpr mice but were much less pathogenic. The possibility that these results were compromised by other factors (the ability of the NODlpr host to decrease the frequency of transferred cells, for example) needs to be considered.
From a physiological point of view, the final effector mechanism leading to irreversible diabetes in the NOD mouse model appears to be mediated by perforin-producing CD8+ T cells (10). NOD mice lacking perforin had islets with infiltrating leukocytes, but diabetes incidence was reduced greatly and was delayed in these animals. Islet damage was seen, however, in perforin-deficient, nondiabetic mice, and it is reasonable to speculate that this resulted from the effects of other molecules such as CD95, tumor necrosis factor receptor I, or cytokine-induced production of nitric oxide. In a small number of animals (16%), these mechanisms even led to diabetes. Our results indicate that CD95 has a minor role in the effector phase of the autoimmune response but give no clue as to its involvement in initiation of disease. CD8+ T cells are essential to initiate infiltration in the NOD mouse pancreas (3), and the studies with perforin-deficient NOD animals have shown that perforin was not required for this to happen. CD95 may be a candidate, but because the immune system of lpr mice is so distorted, it is difficult to conclude anything from these animals, even though they do not develop insulitis.
From the point of view of the experimenter trying to make a death-resistant β cell, each mechanism of β cell killing is important. For example, some perforin-deficient NOD mice developed diabetes (10), and absence of perforin and CD95 had little impact on the rejection of allogeneic islet grafts (37). Encapsulating islets in membranes in an attempt to sequester them from lymphocytes and myeloid cells has not been entirely successful (38). It probably will be necessary to engineer a β cell that lacks death receptors (CD95 and tumor necrosis factor receptor I) and that contains proteins (for example, Bcl-2 homologs or crmA) that help it resist the impact of molecules such as perforin, nitric oxide, and cytokines.
It is apparent that insulin-dependent diabetes mellitus is a multifactorial disease involving both antigen-specific and antigen nonspecific processes. In the NOD mouse model, it is clear that a number of mechanisms can precipitate disease, depending on how the system is manipulated. However, the physiological progression to spontaneous diabetes in NOD animals depends primarily on perforin as the final effector (10). In humans, different effector mechanisms may predominate in different individuals with a similar final outcome, death of all of the β cells and diabetes.
Acknowledgments
We thank Dr. W. R. Heath for helpful discussions, Dr. C. Kurts for help with the CFSE experiment, and Dr. S. Mihajlovic for excellent histological preparations. Tanya Templeton provided invaluable assistance in care of animals and diabetes monitoring. This work was supported by grants from the National Health and Medical Research Council of Australia.
ABBREVIATIONS
- CD95L
CD95 ligand
- CFSE
5,6-carboxy-succinimidyl-fluorescein-ester
- NOD
nonobese diabetic
- FCS
fetal calf serum
References
- 1.Baxter A G, Cooke A. Diabetes Metab Rev. 1995;11:315–335. doi: 10.1002/dmr.5610110403. [DOI] [PubMed] [Google Scholar]
- 2.Tisch R, McDevitt H. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 3.Andre I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang B, Gonzalez A, Benoist C, Mathis D. Eur J Immunol. 1996;26:1762–1769. doi: 10.1002/eji.1830260815. [DOI] [PubMed] [Google Scholar]
- 5.Jarpe A J, Hickman M R, Anderson J T, Winter W E, Peck A B. Reg Immunol. 1990;3:305–317. [PubMed] [Google Scholar]
- 6.Wang Y, Pontesilli O, Gill R G, La Rosa F G, Lafferty K J. Proc Natl Acad Sci USA. 1991;88:527–531. doi: 10.1073/pnas.88.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christianson S W, Shultz L D, Leiter E H. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 8.Hutchings P R, Simpson E, O’Reilly L A, Lund T, Waldmann H, Cooke A. J Autoimmun. 1990;3:101–109. doi: 10.1016/s0896-8411(09)90018-x. [DOI] [PubMed] [Google Scholar]
- 9.Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon J W. J Immunol. 1994;152:2042–2050. [PubMed] [Google Scholar]
- 10.Kagi D, Odermatt B, Seiler P, Zinkernagel R M, Mak T W, Hengartner H. J Exp Med. 1997;186:989–997. doi: 10.1084/jem.186.7.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allison J, Miller J F A P, Sarvetnick N. Cytokines in Autoimmunity. Landes, TX: Medical Intelligence Unit; 1996. pp. 49–75. [Google Scholar]
- 12.Mandrup-Poulsen T. Diabetologia. 1996;39:1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 13.Nagata S. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa M, Yokono K, Nagata M, Hatamori N, Ogawa W, Miki A, Mizoguti H, Baba S. Diabetes. 1991;40:1210–1217. doi: 10.2337/diab.40.9.1210. [DOI] [PubMed] [Google Scholar]
- 15.Ankarcrona M, Dypbukt J M, Brune B, Nicotera P. Exp Cell Res. 1994;213:172–177. doi: 10.1006/excr.1994.1187. [DOI] [PubMed] [Google Scholar]
- 16.Kurrer M O, Pakala S V, Hanson H L, Katz J D. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Augustine, P., Stephens, L. A., Allison, J., Elefanty, A. G., Ekberg, M., Kay, T. W. H. & Harrison, L. C. (1998) Mol. Med., in press. [PMC free article] [PubMed]
- 18.Leithauser F, Dhein J, Mechtersheimer G, Koretz K, Bruderlein S, Henne C, Schmidt A, Debatin K M, Krammer P H, Moller P. Lab Invest. 1993;69:415–429. [PubMed] [Google Scholar]
- 19.Stassi G, Todaro M, Richiusa P, Giordano M, Mattina A, Sbriglia M S, Lo Monte A, Buscemi G, Galluzzo A, Giordano C. Tranplant Proc. 1995;27:3271–3275. [PubMed] [Google Scholar]
- 20.Yamada K, Takane-Gyotoku N, Yuan X, Ichikawa F, Inada C, Nonaka K. Diabetologia. 1996;39:1306–1312. doi: 10.1007/s001250050574. [DOI] [PubMed] [Google Scholar]
- 21.Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M. J Exp Med. 1997;186:1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chervonsky A V, Wang Y, Wong F S, Visintin I, Flavell R A, Janeway C A, Jr, Matis L A. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 23.Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. J Exp Med. 1997;186:613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen P L, Eisenberg R A. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 25.Strasser A, Harris A W, Huang D C, Krammer P H, Cory S. EMBO J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandler P, Fairchild S, Simpson E. J Immunogenet. 1988;15:321–330. doi: 10.1111/j.1744-313x.1988.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 27.Wicker L S, Miller B J, Mullen Y. Diabetes. 1986;35:855–860. doi: 10.2337/diab.35.8.855. [DOI] [PubMed] [Google Scholar]
- 28.Lyons A B, Parish C R. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 29.Allison J, Campbell I L, Morahan G, Mandel T E, Harrison L C, Miller J F A P. Nature (London) 1988;333:529–533. doi: 10.1038/333529a0. [DOI] [PubMed] [Google Scholar]
- 30.Chu J L, Ramos P, Rosendorff A, Nikolic-Zugic J, Lacy E, Matsuzawa A, Elkon K B. J Exp Med. 1995;181:393–398. doi: 10.1084/jem.181.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe D, Suda T, Hashimoto H, Nagata S. EMBO J. 1995;14:12–18. doi: 10.1002/j.1460-2075.1995.tb06970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suda T, Tanaka M, Miwa K, Nagata S. J Immunol. 1996;157:3918–3924. [PubMed] [Google Scholar]
- 33.Theofilopoulos A N, Balderas R S, Gozes Y, Aguado M T, Hang L M, Morrow P R, Dixon F J. J Exp Med. 1985;162:1–18. doi: 10.1084/jem.162.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perkins D L, Michaelson J, Glaser R M, Marshak-Rothstein A. J Immunol. 1987;139:1406–1413. [PubMed] [Google Scholar]
- 35.Bobe P, Bonardelle D, Reynes M, Godeau F, Mahiou J, Joulin V, Kiger N. J Immunol. 1997;159:4197–4204. [PubMed] [Google Scholar]
- 36.Wang J K M, Zhu B, Ju S T, Tschopp J, Marshak-Rothstein A. Cell Immunol. 1997;179:153–164. doi: 10.1006/cimm.1997.1159. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed K R, Guo T B, Gaal K K. Transplantation. 1997;63:951–957. doi: 10.1097/00007890-199704150-00008. [DOI] [PubMed] [Google Scholar]
- 38.Zekorn T D, Horcher A, Mellert J, Siebers U, Altug T, Emre A, Hahn H J, Federlin K. Int J Artif Organs. 1996;19:251–257. [PubMed] [Google Scholar]