

Abstract
Some 50% of human cancers are associated with mutations in the core domain of the tumor suppressor p53. Many mutations are thought just to destabilize the protein. To assess this and the possibility of rescue, we have set up a system to analyze the stability of the core domain and its mutants. The use of differential scanning calorimetry or spectroscopy to measure its melting temperature leads to irreversible denaturation and aggregation and so is useful as only a qualitative guide to stability. There are excellent two-state denaturation curves on the addition of urea that may be analyzed quantitatively. One Zn2+ ion remains tightly bound in the holo-form of p53 throughout the denaturation curve. The stability of wild type is 6.0 kcal (1 kcal = 4.18 kJ)/mol at 25°C and 9.8 kcal/mol at 10°C. The oncogenic mutants R175H, C242S, R248Q, R249S, and R273H are destabilized by 3.0, 2.9, 1.9, 1.9, and 0.4 kcal/mol, respectively. Under certain denaturing conditions, the wild-type domain forms an aggregate that is relatively highly fluorescent at 340 nm on excitation at 280 nm. The destabilized mutants give this fluorescence under milder denaturation conditions.
Keywords: tumor suppressor, denaturation, folding, zinc, protein
The tumor suppressor protein p53 is a sequence-specific transcription factor that functions to maintain the integrity of the genome (1). On its induction in response to DNA damage, p53 promotes cell cycle arrest in G1 phase (2) and apoptosis if DNA repair is not possible (3). Negative regulation occurs by the synthesis and subsequent binding of the oncoprotein Mdm2 to the transactivation domain of p53. This targets it for degradation and ensures that the cellular stability of p53 is low (4, 5). About 50% of human cancers and 95% of lung cancers are associated with mutations in p53. The majority of these map to its core domain, which is responsible for binding DNA (6). The crystal structure of the core domain bound to DNA has been determined (7). A number of the tumorigenic mutants affect residues that contact the DNA, but many are not directly involved in binding and appear to affect the thermodynamic stability of the protein (8, 9).
p53 is a possible target for cancer therapy, including drugs that can stabilize it or using superstable p53 variants that would be suitable for gene therapy applications. There is a lack of quantitative information on the stability of p53 on which to base experiments measuring its change in stability on mutation. Data tend to be restricted so far to measurements of the temperature dependence of transactivation or PAb 1620 binding (8, 9), a monoclonal antibody specific for the native state of wild-type p53 (10). These suggest that p53 is relatively unstable. We find in this study that the core domain denatures irreversibly with temperature, and so the Tm measured by differential scanning calorimetry or spectroscopy cannot be used quantitatively for analyzing structure–activity relationships of p53. We have turned instead to studying the stability of the isolated core domain by using urea-mediated denaturation, which is of proven use for systematic measurements of the effects of mutation on stability (11–14). There are potential problems because of Zn2+ ions being bound to p53 and the resultant complications of various holo- and apo-forms of the native and denatured state. We have found procedures for measuring the equilibria between the holo-denatured and native states, which may be used to measure the free energy of unfolding in water,  .
.
MATERIALS AND METHODS
Gene Subcloning and Mutagenesis.
The portion of the human p53 core domain encoding residues 94–312 was amplified by PCR from the plasmid pT7hp53 (15) by using the oligonucleotides 5′-GGG AAT TCC ATA TGT CAT CTT CTG TCC CTT CCC AGA AAA CCT ACC AG-3′ and 5′-GGG AAT TCA GGT GTT GTT GGG CAG TGC TCG CTT AGT GCT CCC-3′. This was digested with NdeI and EcoRI restriction enzymes and subcloned into the polylinker region of pRSET(A) (Invitrogen). The ligated plasmid was transformed into Escherichia coli strain DH5α. The subsequently recovered plasmid, pRSET(A)-corehp53, was sequenced by using T7 promoter and T7 terminator primers (5′-TAA TAC GAC TCA CTA TAG GG-3′ and 5′-CGT AGT TAT TGC TCA GCG G-3′, respectively). Mutagenesis was performed with inverse PCR or the QuikChange site-directed mutagenesis kit (Stratagene).
Protein Expression and Purification.
Protein was expressed in E. coli strain BL21(DE3), which was grown at 37°C to an OD600 = 1.2 before overnight induction at 25°C with 1 mM isopropyl β-d-thiogalactoside. Cells were harvested by centrifugation and sonicated in 50 mM Tris, pH 7.2/5 mM DTT/1 mM phenylmethylsulfonyl fluoride. Soluble lysate was loaded onto a SP-Sepharose cation exchange column (Pharmacia) and eluted with a NaCl gradient (0–1 M). Further purification was achieved by affinity chromatography with a heparin-Sepharose column (Pharmacia) in 50 mM Tris, pH 7.2/5 mM DTT with a NaCl gradient (0–1 M) for elution. Protein concentration was measured spectrophotometrically by using an extinction coefficient of ɛ280 = 17,130 cm−1⋅M−1 calculated by the method of Gill and von Hippel (16). The accuracy of this method was confirmed by amino acid analysis. Electrospray mass spectrometry showed that the initiator methionine residue is cleaved after translation.
Differential Scanning Calorimetry (DSC).
DSC experiments were performed by using a Microcal VPDSC (Microcal, Amherst, MA) with a cell volume of 0.5 ml. Temperatures from 4 to 42°C or 95°C were scanned at a rate of 60°C/h. Dialyzed samples (7–36 μM protein) were degassed and flushed with argon for 10–15 min on ice. The dialysis buffer was used for baseline scans. A pressure of 25 psi (1 psi = 6.89 kPa) was applied to the cell, and the system was allowed to equilibrate at 4°C for 20 min before scanning. The data were analyzed with Origin software (Microcal).
DNA binding experiments with DSC were carried out in 50 mM Tris, pH 7.2/1 mM DTT. The apparent Tm of p53 core domain, the double-stranded oligonucleotide consensus sequence (22-mer) (7), and the complex of double-stranded DNA–protein in equimolar amounts (7 μM) were determined with and without 0.3 M NaCl in solution. Consensus DNA was synthesized as single-stranded oligonucleotides (5′-ATA ATT GGG CAA GTC TAG GAA A-3′ and 5′-TTT CCT AGA CTT GCC CAA TTA T-3′), which were then annealed for DNA binding studies.
Equilibrium Denaturation.
Denaturation was monitored by fluorescence using an Aminco-Bowman Series 2 luminescence spectrofluorimeter with excitation at 280 nm (bandpass, 4 nm), and this emission scanned from 300 to 370 nm (bandpass, 4 nm). Stock urea solutions were prepared gravimetrically. One-hundred microliters of protein (≈18 μM) in the relevant buffer was added to 800 μl of buffer containing the appropriate concentration of urea. Each sample was incubated at 10°C for at least 5 h before fluorescence was measured with four matched 1-ml cuvettes in thermostatted cuvette holders at the same temperature. The temperature was monitored by a thermocouple in the cell.
The data were fitted to Eq. 1, which assumes a two-state model in which the fluorescence of the folded and unfolded states is dependent on denaturant concentration (11):
 |
1 |
where F is the fluorescence at the given [denaturant], αN and αD are the intercepts, and βN and βD are the slopes of the baselines at low (N) and high (D) denaturant concentrations, respectively, [D] is the concentration of denaturant, [D]50% is the concentration of denaturant at which half of the protein is denatured, m is the slope of the transition, R is the gas constant, and T is the temperature in K. The data were fitted to this equation by nonlinear least squares analysis by using the general curve fit option of the Kaleidagraph program (Abelbeck Software, Reading, PA), which gives the calculated values for individual experimental measurements of m and [D]50% together with their standard errors.
The free energy of denaturation of proteins in the presence of denaturant ΔGD-ND is, to a first approximation, linearly related to the concentration of denaturant (17) (Eq. 2):
 |
2 |
The difference in free energy of denaturation between wild-type and mutant proteins may be calculated from:
 |
3 |
where Δ[D]50% is the difference between the value of [D]50% for wild-type and mutant and <m> is the average value of 〈m〉 (12).
RESULTS
DSC.
Thermal denaturation of p53 core domain was found to be irreversible by using a range of pH values, buffers, and other reagents and so can be used only as a qualitative guide to stability. Irreversible denaturation occurred during a single DSC scan, and the formation of an aggregate was seen in attempts to measure the Tm by circular dichroism. The DSC exotherms show a strong pH dependence with apparent values of Tm of 42°C in 20 mM sodium phosphate, pH 7.4/150 mM NaCl/1 mM DTT and 36°C in 40 mM Mes, pH 6.0/1 mM DTT. DSC was used qualitatively to examine the DNA binding of p53 core domain (Fig. 1). The addition of a 22-mer double-stranded DNA p53 consensus sequence was found to raise the apparent Tm in 50 mM Tris buffer from 42 to 49°C. This result shows all the protein is able to bind DNA, which has a significant stabilizing effect. This effect was reduced in the presence of 300 mM NaCl.
Figure 1.

DNA binding analysis of p53 core domain from DSC. Thermograms were determined in 50 mM Tris, pH 7.2/1 mM DTT of (top trace) double-stranded DNA (dsDNA) oligonucleotide (7 μM) (Tm = 56°C), (middle trace) p53 core domain + dsDNA oligonucleotide equimolar (Tm = 49°C), (bottom trace) p53 core domain (7 μM) (Tm = 42°C) alone. Data are shown without correction for the buffer baseline and are offset for clarity.
Fluorescence Spectra.
On excitation at 280 nm, the single tryptophan residue in denatured p53 fluoresces strongly at 350 nm, whereas it fluoresces very weakly in native p53. The difference in fluorescence is at a maximum at 356 nm. The fluorescence spectra during urea denaturation in phosphate or Tris buffer show a clear isofluorescent point (around 316–321 nm, dependent on spectrofluorimeter and mutant used), suggesting that only two species are present during denaturation (Fig. 2), apart from when aggregation occurs (see below). The isofluorescent point can be used to normalize fluorescence data for small differences in protein concentration or cuvette size. The weaker fluorescence of tyrosine residues at 305 nm decreases on denaturation.
Figure 2.

Fluorescence of p53 core domain in 50 mM sodium phosphate, pH 7.2/5 mM DTT at 25°C on excitation at 280 nm. The native state (N) has a tyrosine emission maximum at 305 nm, which shifts to 310 nm on denaturation. There is a clear tryptophan emission maximum for the denatured state (D) at 350 nm. The tryptophan fluorescence of N is very weak. D and N have the same fluorescence at 321 nm. Aggregated states have a fluorescence maximum at 340 nm; shown is the emission of p53 core domain after being heated to 60°C and cooled to 25°C.
Zinc Binding During Equilibrium Denaturation.
Biochemical studies on the binding of DNA show that DTT is required to maintain p53 in the reduced state (18), so DTT has been used throughout the folding experiments described here. The use of DTT with zinc-binding proteins can create problems, as DTT forms a stable Zn–DTT complex of low solubility (19). But equilibrium measurements on mutants studied here at 10°C and wild type at 25°C appear independent of the concentration of DTT in the range of 40 μM to 40 mM. This suggests that there is no loss of Zn2+ ion during denaturation, with the two large loop elements (loops 2 and 3) binding Zn2+ in the denatured state with a higher affinity than DTT. Direct measurement of Zn2+ by using a spectrophotometric assay with the metallochromic indicator 4-(2-pyridylazo)resorcinol (20) and mercurial-promoted Zn2+ release shows that 1 mol is bound per mol of native core domain and that this remains bound in 5 M urea. The chemical equilibrium is just between the two species at the top of Fig. 3 with no significant accumulation of the other possible equilibrium species.
Figure 3.

Scheme for the equilibria between the native and denatured states of p53 core domain with and without bound zinc. The equilibrium that has been measured is shown by KD-NH.
Aggregation of p53 Core Domain.
During urea-induced denaturation at 37°C, or even 25°C in non-phosphate buffers, there is a transient species that has an emission maximum at 340 nm with an intensity greater than that from the denatured state at 356 nm (Fig. 2). Gel filtration revealed this species to be an aggregate. The 340 nm fluorescence is also observed during the thermal unfolding of the protein or under moderately denaturing conditions (1–2 M urea) in the presence of 1 mM EDTA, which chelates Zn2+. A visible aggregate is formed at increased protein concentrations (≈25 μM); the 340 nm fluorescence is removed on centrifugation. The same fluorescence is observed when mutants of the core domain are subjected to milder denaturation conditions.
Urea Denaturation.
The denaturation of wild-type and mutant p53 core domains was measured from the changes in fluorescence on the addition of urea. A complete emission spectrum was recorded at each concentration of urea so denaturation curves could be analyzed from changes in tryptophan and tyrosine fluorescence, any aggregation could be detected at 340 nm, and the isofluorescent point checked. There was excellent agreement between the changes at 305 nm for tyrosines and 356 nm for the lone tryptophan (which is 25% solvent exposed), showing a cooperative transition. The denaturation curves are entirely reversible at 10°C, identical results being obtained on the renaturation of a denatured sample. Therefore, equilibrium measurements can be made from denaturation curves, which fit to a two-state transition (Fig. 4). Denaturation of wild-type protein is also fully reversible at 25°C, but a standard temperature of 10°C was chosen because some mutants do not denature reversibly at higher temperatures. R175H appears to be at the stability limit for our folding studies because of its degree of degradation on purification. Gel filtration was used as a third column step to purify folded R175H from smaller fragments, although resolution was not complete. The raw data at 356 nm without normalization are shown in Fig. 4 and give a good measurement of [urea]50%. Results of denaturation are presented in Table 1.
Figure 4.

Urea denaturation of wild-type (WT) and mutant p53 core domain monitored by normalized fluorescence emission at 356 nm (data without normalization are shown for R175H; excellent data are obtained in general without normalization). Data are plotted as fraction unfolded and fitted to Eq. 1. Protein was buffered in 50 mM sodium phosphate, pH 7.2/5 mM DTT. An excitation wavelength of 280 nm was used.
Table 1.
Equilibrium denaturation of p53 core domain
| p53 variant* | m, kcal⋅mol−1⋅M−1 | [Urea]50%, M |
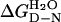 †, kcal⋅mol−1 †, kcal⋅mol−1
|
ΔΔGD-N3M urea†, kcal⋅mol−1 |
|---|---|---|---|---|
| Wt, 25°C (1) | −2.24 ± 0.008 | 2.66 ± 0.01 | 5.96 ± 0.20 | |
| Wt, 10°C (2) | −2.97 ± 0.18 | 3.33 ± 0.01 | 9.76 ± 0.24 | |
| R175H, 10°C (1) | −2.59 ± 0.19 | 2.30 ± 0.02 | 6.75 ± 0.17 | 3.01 ± 0.08 |
| C242S, 10°C (2) | −2.68 ± 0.01 | 2.33 ± 0.01 | 6.82 ± 0.16 | 2.94 ± 0.06 |
| R248Q, 10°C (1) | −2.91 ± 0.15 | 2.67 ± 0.01 | 7.82 ± 0.19 | 1.94 ± 0.05 |
| R249S, 10°C (3) | −3.09 ± 0.09 | 2.66 ± 0.01 | 7.81 ± 0.19 | 1.95 ± 0.04 |
| R273H, 10°C (1) | −3.11 ± 0.10 | 3.21 ± 0.01 | 9.41 ± 0.23 | 0.35 ± 0.04 |
The number of determinations is shown in parentheses. Data are from normalized fluorescence emission at 356 nm (except for R175H). The standard errors of the data are shown. [Urea]50% values can be determined with a high degree of reproducibility but m values are hard to determine accurately, particularly where the [urea]50% value is low (12). Protein was buffered in 50 mM sodium phosphate, pH 7.2/5 mM DTT. Wt, wild type.
The mean m value of 10 urea denaturation curves at 10°C is −2.93 (±0.07) kcal⋅mol−1·M−1, which was used to calculate 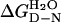 and ΔΔGD-N3M urea from Eq. 2 and Eq. 3, respectively. The values of ΔΔGD-N3M urea are calculated for 3 M urea because there is only a short extrapolation of the data and so the standard error is small. There are higher standard errors at 0 M urea. Values of ΔΔGD-N tend to change only slightly with temperature.
and ΔΔGD-N3M urea from Eq. 2 and Eq. 3, respectively. The values of ΔΔGD-N3M urea are calculated for 3 M urea because there is only a short extrapolation of the data and so the standard error is small. There are higher standard errors at 0 M urea. Values of ΔΔGD-N tend to change only slightly with temperature.
DISCUSSION
We have found conditions to measure the reversible denaturation of p53 core domain and many of its mutants with urea denaturation. Importantly, a single Zn2+ ion is bound throughout the reaction, because its loss would generate denaturation curves that are very difficult to analyze because of changes in molecularity. Aggregation was observed under certain conditions, monitored from fluorescence at 340 nm. We suggest the 340 nm fluorescence is the signature of the aggregated state of p53 core domain. At 10°C, aggregation is avoided and unfolding is entirely reversible with denaturation curves starting from folded or unfolded protein overlaying to give the same m and [urea]50% values (Eq. 1), thus allowing the free energy of unfolding ΔGD-N, to be calculated. Most proteins are stable by some 5–15 kcal⋅mol−1 at 25°C (21). Here, we show that the isolated core domain is of moderate thermodynamic stability (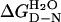 = 6.0 kcal⋅mol−1, 25°C). The domain will be even less stable at 37°C so only small changes in stability will lead to loss of function.
= 6.0 kcal⋅mol−1, 25°C). The domain will be even less stable at 37°C so only small changes in stability will lead to loss of function.
Mutants of p53 Core Domain Are Destabilized.
Mutants of p53 core domain, corresponding to known “hot spots” associated with tumors, were constructed and their stabilities were determined (Fig. 5). Some of the residues mutated interact directly with DNA whereas others are part of the protein scaffold (“structural” mutants).
Figure 5.

Molscript (22) picture of p53 core domain. Residues mutated are labeled and shown as stick models as is Trp-146, which was used as a fluorescent probe for denaturation.
The stabilities fall into three classes. The DNA-binding mutant R273H is of similar stability to wild type, whereas C242S and R175H, which surround the Zn2+-binding site, are destabilized by 2.9 and 3.0 kcal⋅mol−1, respectively. R248Q and R249S are of intermediate stability. These results are qualitatively consistent with studies on antibody binding (8, 9). R175H reacts strongly with an antibody specific for the denatured p53 and does not react with an antibody specific for the folded state of the domain; and, conversely, R273H has the opposite specificity.
The distinction between structural (R249S) and DNA contact mutations (R248Q) is shown here to be less straightforward than previously thought. R248Q was presumed to have no role in stability yet fits both categories and demonstrates the general trend that the degree of destabilization is more severe the closer residues lie to the Zn2+-binding site. A spectrophotometric assay (20) shows that C242S retains a bound Zn2+ ion throughout denaturation despite the loss of one cysteine ligand. The replacement of arginine with histidine at residue 175 creates the possibility of competition between Cys-176 and His-175 for Zn2+ ligation, which could create a His2-Cys2 motif. The results show the importance of residues in loops 2 and 3 and in particular those at the Zn2+-binding site. These residues have been shown to form a network of salt bridges, hydrogen bonds, and van der Waals contacts, which help present DNA binding residues for their major and minor groove binding sites.
Implications for Drug Therapy.
Although the mutants R175H, C242S, R248Q, R249S, and R273H are less stable (Table 1), they do retain cooperative two-state unfolding transitions (Fig. 4), demonstrating defined stable structures at 10°C. The degree of destabilization of mutants reported here is consistent with protein engineering studies of other proteins (12). The changes in stability are sufficiently small to allow possible therapeutic use of small molecules to rescue p53 function by stabilizing it. Any protein-stabilizing drug for effective cancer treatment must, however, also create the correct geometry at the DNA binding surface as many mutants do not show wild-type specificity or binding levels even when folded at low temperature (9).
Acknowledgments
We thank Dr. Mark Bycroft for helpful advice. This work was supported by the Cancer Research Campaign of the United Kingdom. J.H. holds a Kekulé scholarship, Fonds der Chemischen Industrie, Germany, B.S.D. is a recipient of a Burroughs Wellcome Fund Hitchings–Elion Fellowship, and P.V.N. is a postdoctoral fellow of the Medical Research Council of Canada.
ABBREVIATION
- DSC
differential scanning calorimetry
References
- 1.Lane D P. Nature (London) 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 3.Polyak K, Xia Y, Zweier J L, Kinzler K W, Vogelstein B. Nature (London) 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A, Shkedy D, Oren M, Bercovich B. J Biol Chem. 1994;269:9582–9589. [PubMed] [Google Scholar]
- 5.Haupt Y, Maya R, Kazaz A, Oren M. Nature (London) 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 6.Pavletich N P, Chambers K A, Pabo C O. Genes Dev. 1993;7:2556–2564. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- 7.Cho Y, Gorina S, Jeffrey P D, Pavletich N P. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 8.Rolley N, Butcher S, Milner J. Oncogene. 1995;11:763–770. [PubMed] [Google Scholar]
- 9.Friedlander P, Legros Y, Soussi T, Prives C. J Biol Chem. 1996;271:25468–25478. doi: 10.1074/jbc.271.41.25468. [DOI] [PubMed] [Google Scholar]
- 10.Milner J, Cook A C, Sheldon M. Oncogene. 1987;1:453–455. [PubMed] [Google Scholar]
- 11.Clarke J, Fersht A R. Biochemistry. 1993;32:4322–4329. doi: 10.1021/bi00067a022. [DOI] [PubMed] [Google Scholar]
- 12.Serrano L, Kellis J T, Jr, Cann P, Matouschek A, Fersht A R. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 13.Shortle D, Stites W E, Meeker A K. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 14.Santoro M M, Bolen D W. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 15.Midgley C A, Fisher C J, Bartek J, Vojtesek B, Lane D, Barnes D. J Cell Sci. 1992;101:183–189. doi: 10.1242/jcs.101.1.183. [DOI] [PubMed] [Google Scholar]
- 16.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 17.Pace C N. Methods Enzymol. 1986;131:266–279. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 18.Rainwater R, Parks D, Anderson M E, Tegtmeyer P, Mann K. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornell N W, Crivaro K E. Anal Biochem. 1972;47:203–208. doi: 10.1016/0003-2697(72)90293-x. [DOI] [PubMed] [Google Scholar]
- 20.Hunt J B, Neece S H, Ginsburg A. Anal Biochem. 1985;146:150–157. doi: 10.1016/0003-2697(85)90409-9. [DOI] [PubMed] [Google Scholar]
- 21.Fersht A R, Serrano L. Curr Opin Struct Biol. 1993;3:75–83. [Google Scholar]
- 22.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]