

Abstract
DNA and other biopolymers differ from classical polymers because of their torsional stiffness. This property changes the statistical character of their conformations under tension from a classical random walk to a problem we call the “torsional directed walk.” Motivated by a recent experiment on single lambda-DNA molecules [Strick, T. R., Allemand, J.-F., Bensimon, D., Bensimon, A. & Croquette, V. (1996) Science 271, 1835–1837], we formulate the torsional directed walk problem and solve it analytically in the appropriate force regime. Our technique affords a direct physical determination of the microscopic twist stiffness C and twist-stretch coupling D relevant for DNA functionality. The theory quantitatively fits existing experimental data for relative extension as a function of overtwist over a wide range of applied force; fitting to the experimental data yields the numerical values C = 120 nm and D = 50 nm. Future experiments will refine these values. We also predict that the phenomenon of reduction of effective twist stiffness by bend fluctuations should be testable in future single-molecule experiments, and we give its analytic form.
The theory of random walks is one of the most fundamental problems in statistical mechanics, with applications throughout physics, biology, and even finance. The discovery that polymer conformations afford a concrete realization of this mathematical problem, and the understanding that rubber elasticity is inherently an entropic phenomenon, marked the birth of polymer physics (1). Remarkably, it recently has become possible to apply minuscule forces to single molecules of DNA in solution and observe their extension (2). Besides allowing a detailed confirmation of the directed random walk model of entropic elasticity, these experiments allow direct physical measurement of microscopic (nanometer-scale) mechanical properties of DNA relevant to its function, by using mesoscopic (micron-scale) apparatus. Two linear elastic parameters of DNA now have been measured in this way: the bend persistence length A and the intrinsic-stretch modulus γ (3–9).
DNA and other stiff biopolymers differ from classical polymers, however, in that they exhibit torsional as well as bend stiffness. Thus their conformations reflect not a classical directed walk but a new fundamental problem: the “torsional directed walk” (TDW), whose random variables are the direction of each step relative to its predecessor, together with a relative axial twist. In this paper we will formulate the version of the TDW appropriate to DNA, solve it analytically in a regime appropriate to a recent experiment (10), and show that the model quantitatively fits the data over a wide range of applied forces (Fig. 1). Some of these results were announced in ref. 11; related work on the scaling limit of the TDW appeared in ref. 12. Besides being transparent, analytic formulae permit systematic least-squares fitting to experimental data. We fit to obtain three microscopic elastic constants: the twist persistence length C, bend persistence length A, and intrinsic twist-stretch coupling D (13–15). Because A is known independently we have a check on the model. The experiment is not sensitive to the other allowed linear-elastic constants such as twist-bend coupling (16).
Figure 1.

Relative extension of lambda-DNA versus applied force f and overtwist σ. From top to bottom, the curves are at fixed force 8.0, 1.3, 0.8, 0.6, 0.3, and 0.1 pN. The dots are experimental data from figure 3 of ref. 10, excluding values of f, σ where the DNA is known to denature by strand separation. Open dots are outside the range of validity of the phantom chain model and were not used in the fit. The corresponding points from figure 2 of ref. 10 also were used in the fit (not shown), for a total of 49 points. The lines are our theoretical predictions after fitting to A, C, and D (see text).
We find that the existing data (10) yield A = 49 nm, C = 120 nm, and D = 50 nm; future experiments will refine these values when fit to our formula. Many authors have sought to extract the value of C from both cyclization experiments and fluorescence depolarization (17–20). A key point of this paper is that the force regime we study is free from some vexing physical and mathematical difficulties that have helped make the determination of C from these experiments controversial. In particular, we can use a continuum model with no need for the short-length cutoff required to make Monte Carlo calculations tractable (ref. 21; C. Bouchiat and M. Mézard, http://xxx.lanl.gov/abs/cond-mat/9706050). Our value for D is similar to within the large errors to recent estimates (13–15).
We also give a simple analytical prediction for the reduction of effective twist stiffness by bend fluctuations. This renormalization may explain why some other determinations of C give lower values than ours. Its existence was appreciated long ago by Shimada and Yamakawa (23), but a clear experimental test has hitherto not been possible (see also ref. 24). Although this effect is only marginally visible in the extant data, again future experiments should be able to test our prediction by checking the dependence of Ceff on the applied force (see below).
C. Bouchiat and M. Mézard (see above-mentioned web site) recently have addressed several overlapping issues. They independently obtained formulae equivalent to our Eqs. 3 and 4 below. We comment on their approach in Discussion.
Experiment
In the experiment of ref. 10, λ-DNA in 10-mM phosphate buffer was bound to a wall at one end and a magnetic bead at the other, with bonds that did not permit free pivoting. Constraining the orientation of the bead with an applied magnetic field thus constrained the orientation of the DNA strand at its end (Fig. 2). Because the bead was too large for the DNA to loop around it, this procedure effectively fixed the total Link of the circuit consisting of the DNA plus a fixed imaginary closing path. Rotating the applied magnetic field then allowed the authors to freeze the Link to any desired value and find the extension Z for various values of the applied stretching force f. Alternatively, the force could be held fixed while the Link was varied, as in Fig. 1 above. The intensive strain variable describing Link is the relative overtwist σ ≡ ΔLk⋅(3.6 nm/L), where L = 16,400 nm is the total contour length.
Figure 2.

Schematic of experiment. The magnetic bead imposes relative overtwist σ and stretching force f.
Subsequent work showed that the DNA undergoes structural transition or strand separation for high applied stresses, roughly f > 0.4 pN and σ < −0.01 or σ > 0.03 (D. Bensimon, personal communication). The loss of twist rigidity is also clearly visible in the curves in ref. 10. We have omitted such points from Fig. 1. Also, DNA undergoes a dramatic overstretching transition at around 60 pN (7, 8); all the data discussed here concern forces f ≤ 8 pN.
Physical Picture
In this section we will describe qualitatively the physics behind the analysis of the next section.
We will begin with a picture of DNA as a thin cylindrical elastic rod of fixed contour length L; below we will discuss corrections reflecting the more detailed architecture of the molecule. The conformations of such a rod under an applied tension are controlled by the elastic energy functional†
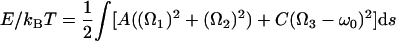 |
1 |
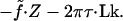 |
Here s is arc length, Ω1,2(s) are bending strains, Ω3 − ω0 is twist strain, A, C are the bend and twist persistence lengths, f̃ ≡ f/kBT, and ω0 ≡ 2π/3.6 nm is the unstressed molecule’s helix density. τ is a dimensionless torque variable whose value we will choose to obtain the required overtwist 〈Lk〉 = (1 + σ)Lω0/2π.
In the absence of thermal fluctuations, a stretched elastic rod remains straight as we apply increasing torque to the ends, then buckles at a critical value of torque, which increases with the applied stretching force (25). Below τcrit = 2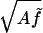 the rod twists uniformly with overtwist σ = τ/ω0C. The end-to-end distance Z of the rod does not change at all for applied overtwist less than the critical value τcrit, because the rod remains a straight line of constant contour length.
the rod twists uniformly with overtwist σ = τ/ω0C. The end-to-end distance Z of the rod does not change at all for applied overtwist less than the critical value τcrit, because the rod remains a straight line of constant contour length.
Thermal fluctuations change this picture completely. The rod is never straight; every Fourier mode of its shape is excited in accordance with the equipartition theorem of statistical physics, so the net length Z is always less than L. The applied tension suppresses those fluctuations of wavenumber smaller than q0 ≡ 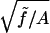 , with the dominant contribution to the shortening coming from q ≈ q0. Now when we apply torque below the critical value, the fluctuations with the same helical sense as the torque get pushed closer to instability and hence grow, whereas those with the opposite helical sense are suppressed. Thus the distribution of rod shapes responds to external torque even below the buckling threshold τcrit. Hence the rod can store imposed Link excess either in Twist, as before, or in a change of average Writhe, and so we will find σ = τ/ω0Ceff with a reduced (or “or renormalized”) effective twist stiffness Ceff. Because the effect depends on bend fluctuations we may expect (Ceff)−1 = C−1 + (A⋅j(Af/kBT))−1, where j−1(x) is some function vanishing at large x. (We will find below that j(x) = 4
, with the dominant contribution to the shortening coming from q ≈ q0. Now when we apply torque below the critical value, the fluctuations with the same helical sense as the torque get pushed closer to instability and hence grow, whereas those with the opposite helical sense are suppressed. Thus the distribution of rod shapes responds to external torque even below the buckling threshold τcrit. Hence the rod can store imposed Link excess either in Twist, as before, or in a change of average Writhe, and so we will find σ = τ/ω0Ceff with a reduced (or “or renormalized”) effective twist stiffness Ceff. Because the effect depends on bend fluctuations we may expect (Ceff)−1 = C−1 + (A⋅j(Af/kBT))−1, where j−1(x) is some function vanishing at large x. (We will find below that j(x) = 4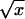 .)
.)
The suppression of some fluctuations and enhancement of others also will affect the end-to-end length Z. These effects cancel to linear order in σ because the elastic energy (Eq. 1) does not break inversion symmetry. We do, however, expect an effect to 𝒪(σ2); indeed this is the dominant feature of the data in Fig. 1. As the force decreases the effect increases, as seen in the increasing curvature of the curves in the figure.
Below we will derive explicit formulas embodying the qualitative arguments in the above two paragraphs.
In Eq. 1 we have neglected any self-avoidance effects; these would appear as interactions between rod elements distant in s. In the usual directed walk this is not a serious omission: the crossover to self-avoiding-walk scaling occurs only for chain lengths much longer, and forces much smaller, than those encountered in DNA. In the torsional directed walk we must be more careful, because the linking number appearing in Eq. 1 is undefined when the chain crosses itself. Physically the problem is that the phantom chain can form a loop, pass through itself, and in the process lose a unit of Link: the phantom torsional chain cannot support any imposed torque. If we seek an equilibrium at nonzero τ we thus must expect to find mathematical pathologies; they will arrive in due course. A related problem is that the statistical sum for the phantom torsional chain includes all knotted configurations, an error with noticeable effects (26).
One approach to this problem is to introduce realistic self-avoidance and knot rejection into Eq. 1 (21, 26). The resulting nonlocal model requires numerical Monte Carlo solution, and the results depend on the details of the chain interaction chosen. From the physical picture, however, it is clear that at high enough applied tension f the problematic loops and knots will be so rare as to be negligible: the chain remains nearly straight, and we may use Eq. 1 without modification. We will find below the precise condition to be in this regime; it corresponds to the solid dots in Fig. 1.
Eq. 1 also neglects any effects of rod anisotropy. For example, bending into the major groove (“roll”) is easier than bending in the perpendicular direction (“tilt”); less obvious is an allowed twist-bend coupling (16). Such anisotropies can lead to chiral effects, for example an asymmetry between σ and −σ, but their effects on Fig. 1 are negligible (unpublished work). Indeed we only expect the helical pitch to affect entropic elasticity when the dominant wavenumber q0 approaches ω0, i.e., at unattainably large forces.‡
Instead the slight asymmetry visible in Fig. 1 has its origin in the intrinsic stretch elasticity of a chiral rod, which gives Z a contribution linear in σ and independent of f (14, 15). Stretching with associated unwinding occurs in vivo when the protein RecA binds to DNA, a step in homologous recombination (27, 28). In the experiment of ref. 10 the effect should be masked by the entropic σ2 term for small force, emerging when the latter is suppressed at high force, exactly as seen in Fig. 1.
Calculation
We now sketch a calculation embodying the above physical picture (unpublished work). We introduce three local configuration variables: a unit vector t(s) describing the tangent to the chain, and an angle ζ(s) for the remaining torsional degree of freedom. We take the applied force along the z direction and orient the chain so that t = z in equilibrium. To define ζ(s) we use Fuller’s local formula for the Writhe of a curve whose tangent never points along the −z axis (29, 30): Wr = 1/2π ∫ ds(t × dt/ds)⋅z/(1+t⋅z). Combined with White’s theorem that 2πWr + ∫ Ω3ds is a topological invariant, we see that Ω3 + (t × dt/ds)⋅z/(1 + t⋅z) must be a total derivative; we will call this quantity ω0 + dζ/ds and eliminate Ω3(s) in favor of ζ(s).§ The advantage of this choice is that Eq. 1 is now quadratic in ζ, which may be summarily eliminated.
It proves convenient to define dimensionless quantities K ≡ 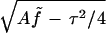 and s̄ ≡ Ks/A; we then find
and s̄ ≡ Ks/A; we then find
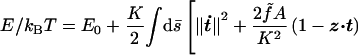 |
2 |
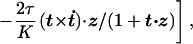 |
where E0 ≡ −L(f̃ + τ2/2C) and dot denotes d/ds̄. The second term defines a nonlinear fluctuation problem, which we will expand in powers of 1/K. From its partition function 𝒵(f, τ) we may then extract the extension and excess link as
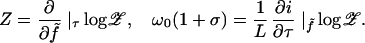 |
3 |
We will use the second of these to solve for τ(f, σ), then substitute into the first to get the desired extension Z(f, σ).
To find 𝒵 we adapt the standard trick used in the wormlike chain (6, 31): for long chains 𝒵 approaches the unnormalized correlation function of t. Holding t(0) fixed, this correlator ψ(t, s) obeys the Schrödinger-like equation ψ̇ = −Hψ, where (http://xxx.lanl.gov/abs/cond-mat/9706050) (unpublished work)
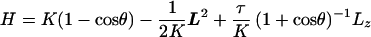 |
4 |
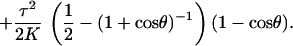 |
Here cos θ = t⋅z, L2 is the angular part of the Laplace operator, and Lz is the azimuthal derivative. The main novelty of this derivation is the presence of first-order derivatives in Eq. 2 when τ ≠ 0, leading to the τ2 terms in K and in Eq. 4. The asymptotic value of 𝒵 is then controlled by the lowest (“ground-state”) eigenvalue λ0 of Eq. 4 via 𝒵 ∝ e−E0−λ0L.
Unfortunately Eq. 4 has no ground state for any nonzero τ, because of its singularity at θ = π! This unphysical pathology was predicted in the previous section; its mathematical origin is the breakdown of Fuller’s formula when t = −z. To see the connection to the physical discussion, note that the unphysical Link-dropping process in the phantom torsional chain necessarily involves the tangent t(s) passing through −z at some intermediate point. As discussed above, a physically meaningful and analytically tractable resolution to the problem is to restrict attention to large f. We can then solve Eq. 4 in perturbation theory about θ = 0, where the problem is invisible, provided the perturbative ground state value is smaller than the “tunneling barrier” of Eq. 4.¶ Imposing this condition and K2 > 2 selects the solid dots in Fig. 1.
The perturbative solution of Eq. 4 gives the ground state eigenvalue λ0 = 1 − 1/4K − 1/64K2 ⋯, so Eq. 3 gives σ = τ/ω0 (C−1 + (4KA)−1 + 𝒪(K−3)), Z/L = 1 − (2K)−1 (1 + 1/64K2 + 𝒪(K−3)). The leading-order approximations to these formulae were announced in ref. 11. Solving by iteration gives the torque
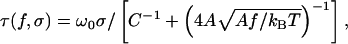 |
5 |
plus corrections of 𝒪(K−3). Eq. 5 displays the promised renormalization of twist stiffness by bend fluctuations. Although direct torque measurements are not currently possible, this effect nevertheless enters the force curves because τ enters Z/L.
Assembling the pieces gives our theoretical prediction for the force curve: the relative extension Z/L is
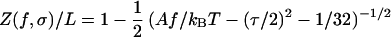 |
6 |
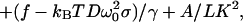 |
with τ from Eq. 5. We have improved the formula for Z/L given above by summing perturbation theory and including a small finite-length correction (unpublished work). We also introduced the intrinsic-stretch terms mentioned in the previous section (6).
In Eqs. 5 and 6 the parameters ω0, L are known, and we use the value γ = 1,100 pN for the linear stretch constant obtained from higher-force experiments (9). This leaves A, C, and D, which we fit to the experimental data after the cuts described above. Fig. 1 shows that a single choice of A = 49 nm, C = 120 nm, D = 50 nm fits all the curves.‖ We note that the fit value of A is consistent with the experiment of Wang et al. (9). Another nontrivial check is that several points just outside our accepted set, not used in the fit (open symbols in Fig. 1), nevertheless lie on our theoretical curves.
Discussion
Our value for C is larger than previous determinations. Cyclization experiments give values like C = 85 nm, whereas fluorescence polarization anisotropy (FPA) gives still smaller values (17–20, 23, 24). Significantly, experiments on short (<100 nm) DNA yields torsion constants about 50% larger than those for longer strands (33). It is tempting to speculate that the difference is because of the reduction of effective twist stiffness derived in the previous section. Indeed, neglecting this renormalization by replacing Eq. 5 with the naïve τ = ω0σ/Ceff and fitting the data gives the value Ceff = 98 nm, closer to earlier determinations than our true microscopic value of C. More generally, equilibrium measurements, like the one reported here, are far more straightforward to interpret than dynamical measurements like FPA.**
Actually, our seemingly large value of C/A could have been anticipated. Random natural bends in DNA reduce the net persistence length at zero applied tension to a value significantly below the value implied by the elastic stiffness alone (34). A similar effect reduces the effective A measured in stretching experiments, but not C (P.N., unpublished work). Estimates for the true elastic contribution to the bend persistence length (the “dynamic persistence length” Ad) range from 80 nm to as much as 210 nm (22, 35, 36), and so the elastic C/Ad could be unity or even greater.
Bouchiat and Mézard (http://xxx.lanl.gov/abs/cond-mat/9706050) recently have analyzed the f = 0.1 pN curve. Because the 0.1 pN curve is entirely outside the range of validity of the phantom chain model, however, they were obliged to address the unphysical pathology of Eq. 4, both by Monte Carlo simulation and by introducing a new short-scale cutoff. The new cutoff introduces a new unknown parameter into the theory, and moreover does not correspond in a simple way to the actual physics of self-avoidance. They found an impressive match to the f = 0.1 pN curve by using C = 75 nm, and relative insensitivity to their choice of cutoff for |σ| < 0.015. Nevertheless we believe our value of C better describes the experiment, because we simultaneously fit to several different values of f.
In this paper we have introduced an abstract statistical-mechanics problem, the torsional directed walk. We solved it in a simple regime and found a clean confirmation in the experiment of ref. 10 with three adjustable parameters, the nanometer-scale elastic constants of DNA. We also predicted quantitatively the reduction of twist stiffness caused by bend fluctuations; a definitive test of Eq. 5 must await further experiments.
Acknowledgments
We thank B. Fain, R. D. Kamien, T. C. Lubensky, J. Marko, C. O’Hern, J. Rudnick, J. M. Schurr, and M. Zapotocky for helpful discussions, C. Bouchiat and M. Mézard for correspondence, and D. Bensimon and V. Croquette for supplying us with experimental details and the numerical data from ref. 10. This work was supported in part by National Science Foundation Grant DMR95-07366. J.D.M. was supported in part by a Fonds pour la Formation de Chercheurs et l’Aide à la Recherche Graduate Fellowship from the government of Quebec.
Note added in proof:
Additional details of this experiment can be found in ref. 37.
Footnotes
Our notation is similar to ref. 16. Throughout we will neglect sequence dependence. In the force regime in question we expect linear rod elasticity to be a good approximation. Higher-order bend elasticity effects are expected to be suppressed by powers of the rod radius (1 nm), which is much smaller than any other length scale. Indeed a linear-elastic model, the “extensible worm-like chain,” describes accurately the extension of torsionally unconstrained DNA up to forces greater than those considered here, with an intrinsic stretch modulus more than 100 times greater than the forces of interest to us (9).
Certainly the omitted anisotropic couplings also will renormalize the constants A, C in Eq. 1 (unpublished work). The model (Eq. 1) is to be regarded as coarse-grained to the scale of the helix pitch.
The leading-order perturbative formulas can be obtained directly, without appeal to White’s formula (11). A more elegant approach takes the configuration variables to be a 3 × 3 rotation matrix; the torque term then takes the form −τ ∫ ds[Ω3 + Ω̌3]/(1 + t⋅z), where Ω̌i are the space-fixed angular velocities of a rigid body (unpublished work).
To justify perturbation theory itself we note that it gives an excellent approximation to the exact solution of the worm-like chain (6) when K > 1, as may be expected from the form of the leading anharmonic correction below. Nonperturbative effects in the variational approach to the worm-like chain (6) are also small when K > 1. Note that our data cuts also eliminate the region σ > 2π/Cω0 where plectoneme formation (and hence large self-avoidance effects) is expected (32). Raising our threshold on K selected fewer points with little effect on our result.
The variance of the data from our curves is σZ/L = 0.013, comparable to the visible scatter in the data. The formal covariances for A, C, D correspond to very small errors; in practice the fit is visibly worse for C outside the range 70 < C < 150. Omitting either the f/γ or the −Dσ/γ terms makes a poorer fit, as does replacing Eq. 5 by the naïve τ = ω0σ/Ceff with constant Ceff.
An independent check of our measured value of the twist-stretch coupling D is not available, but it is interesting to compare to the situation with RecA. Binding to RecA unwinds it to σ = −0.52 and stretches it by 0.54 times its natural length. Unwinding DNA slightly at constant tension lengthens it by DσkBTω02/γ (14, 15). Although σ = −0.52 is outside the domain of linear elasticity, applying the formula gives an extension of 0.33; much of the extension is explained by the unwinding without requiring additional tension. This argument at best confirms that our value of D is not unreasonable.
References
- 1.Flory P J. Statistical Mechanics of Chain Molecules. New York: Interscience; 1969. [Google Scholar]
- 2.Austin R H, Brody J P, Cox E C, Duke T, Volkmuth W. Physics Today. 1997;50:32–38. and references therein. [Google Scholar]
- 3.Smith S B, Finzi L, Bustamante C. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- 4.Bustamante C, Marko J F, Siggia E D, Smith S. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 5.Vologodskii A V. Macromolecules. 1994;27:5623–5625. [Google Scholar]
- 6.Marko J F, Siggia E D. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 7.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J-L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 8.Smith S, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 9.Wang M D, Yin H, Landick R, Gelles J, Block S M. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strick T R, Allemand J-F, Bensimon D, Bensimon A, Croquette V. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 11.Nelson, P. (1997) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 12.Moroz J D, Kamien R D. Nucl Phys. 1997;B506:695–710. [Google Scholar]
- 13.Marko J F. Europhys Lett. 1997;38:183–188. [Google Scholar]
- 14.Kamien R D, Lubensky T C, Nelson P, O’Hern C S. Europhys Lett. 1997;38:237–242. [Google Scholar]
- 15.Kamien, R. D., Lubensky, T. C., Nelson, P. & O’Hern, C. S. (1998) Eur. Phys. J., in press.
- 16.Marko J F, Siggia E D. Macromolecules. 1994;27:981–988. [Google Scholar]
- 17.Record M, Mazur S, Melancon P, Roe J, Shaner S, Unger L. Annu Rev Biochem. 1981;50:997–1024. doi: 10.1146/annurev.bi.50.070181.005025. [DOI] [PubMed] [Google Scholar]
- 18.Hagerman P G. Ann Rev Biophys Biophys Chem. 1988;17:265–286. doi: 10.1146/annurev.bb.17.060188.001405. [DOI] [PubMed] [Google Scholar]
- 19.Allison S, Austin R, Hogan M. J Chem Phys. 1989;90:3843–3854. [Google Scholar]
- 20.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 21.Marko J F, Vologodskii A. Biophys J. 1997;73:123–132. doi: 10.1016/S0006-3495(97)78053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song L, Schurr J M. Biopolymers. 1990;30:229–237. doi: 10.1002/bip.360300302. [DOI] [PubMed] [Google Scholar]
- 23.Shimada J, Yamakawa H. Macromolecules. 1984;17:689–698. [Google Scholar]
- 24.Shore D, Baldwin R L. J Mol Biol. 1983;170:957–981. doi: 10.1016/s0022-2836(83)80198-3. [DOI] [PubMed] [Google Scholar]
- 25.Love A E H. A Treatise on the Mathematical Theory of Elasticity. 4th Ed. New York: Dover; 1944. [Google Scholar]
- 26.Vologodskii A V, Cozzarelli N R. Annu Rev Biophys Biomol Struct. 1994;23:609–43. doi: 10.1146/annurev.bb.23.060194.003141. and references therein. [DOI] [PubMed] [Google Scholar]
- 27.Stasiak A, DiCapua E. Nature (London) 1982;299:185–186. [Google Scholar]
- 28.Stasiak A, DiCapua E, Koller T. Cold Spring Harbor Symp Quant Biol. 1983;47:811–820. doi: 10.1101/sqb.1983.047.01.093. [DOI] [PubMed] [Google Scholar]
- 29.Fuller F B. Proc Natl Acad Sci USA. 1978;75:3357–3561. doi: 10.1073/pnas.75.8.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fain B, Rudnick J, Östlund S. Phys Rev E. 1996;55:7364–7368. [Google Scholar]
- 31.Grosberg A Yu, Khokhlov A R. Statistical Mechanics of Macromolecules. New York: Am. Instit. Physics Press; 1994. [Google Scholar]
- 32.Marko J F. Phys Rev E. 1997;55:1758–1772. [Google Scholar]
- 33.Heath P J, Gebe J A, Allison S A, Schurr J M. Macromolecules. 1996;29:3583–3596. [Google Scholar]
- 34.Trifonov E, Sussman J L. Proc Natl Acad Sci USA. 1980;77:3816–3820. doi: 10.1073/pnas.77.7.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bednar J, Furrer P, Katritch V, Stasiak A Z, Dubochet J, Stasiak A. J Mol Biol. 1995;254:579–591. doi: 10.1006/jmbi.1995.0640. [DOI] [PubMed] [Google Scholar]
- 36.Allison S, Austin R, Hogan M. J Chem Phys. 1989;90:3843–3854. [Google Scholar]
- 37.Strick, T. R., Allemand, J.-F., Bensimon, D. & Croquette, V. (1998) Biophys. J., in press. [DOI] [PMC free article] [PubMed]