

Abstract
Titin is a very large alternatively spliced protein that performs multiple functions in heart and skeletal muscle. A rat strain is described with an autosomal dominant mutation that alters the isoform expression of titin. While wild type animals go through a developmental program where the 3.0 MDalton N2B becomes the major isoform expressed by two to three weeks after birth (~85%), the appearance of the N2B is markedly delayed in heterozygotes and never reaches more than 50% of the titin in the adult. Homozygote mutants express a giant titin of the N2BA isoform type (3.9 MDalton) that persists as the primary titin species through ages of more than one and half years. The mutation does not affect the isoform switching of troponin T, a protein that also is alternatively spliced with developmental changes. The basis for the apparently greater size of the giant titin in homozygous mutants was not determined, but additional length was not due to inclusion of sequence from larger numbers of PEVK exons or the Novex III exon. Passive tension measurements using isolated cardiomyocytes from homozygous mutants showed that cells could be stretched to sarcomere lengths greater than 4 µm without breakage. This novel rat model should be useful for exploring the potential role of titin in the Frank-Starling relationship and mechano-sensing/signaling mechanisms.
Keywords: titin, alternative splicing, isoforms, passive tension, heart
1. Introduction
Titin [1], also known as connectin [2], is an extremely large protein (subunit weight exceeding 3 MDa) found primarily in cardiac and skeletal muscles. The protein has been implicated in a number of functions, including protection of the sarcomere against overstretch, centering the A band between the Z lines in the sarcomere, sarcomere assembly, serving as a template for length determination of the thick filaments, the source of passive tension, lengthening force from very short sarcomere lengths, phosphorylation of telethonin, control of protein turnover, binding more than 20 other proteins, and participation in signaling pathways in response to muscle stretch (see [3–7] for reviews). There is a single titin gene in higher eukaryotes, but multiple isoforms are expressed due to alternative splicing[8–10]. The heart contains two major isoform classes, N2B and N2BA, with the former being shorter in length [9]. The ratio of amounts of these two classes varies between species [11] with small mammals having a predominance of the smaller N2B isoform while large mammals, including humans, have a more equal representation of both classes. The isoform ratio changes in rats during development starting with a single large N2BA species at the embryonic stage and proceeding to the adult pattern by 20 days after birth [12–14]. The developmental transformation appears to vary between species [15]. The ratio of the N2BA to N2B isoforms affects passive tension in the heart with muscle containing higher N2BA proportions exhibiting lower tension at a given sarcomere length. The isoform ratio was shown to be altered by hypertension in the rat [16] and, artificially, by chronic pacing in the dog [17]. A genome-wide linkage scan found that exercise induced changes in cardiac stroke volume was linked to the titin gene chromosome region [18]. Human cardiac N2BA to N2B ratios have been found to be modified in hearts from individuals with congestive heart failure [19] and dilated cardiomyopathy [20–22]. It is currently unclear whether these isoform changes are beneficial to health or maladaptive [23].
The importance of alternative splicing for creating protein diversity has been increasingly recognized, and it has been estimated that 74% of all human genes give rise to alternatively spliced transcripts [24]. The mechanisms controlling splicing have been elusive, and little is known about the factors controlling transcript levels. Factors for determining titin isoform expression are currently unknown. However, an initial description of a strain of rats that showed altered titin isoform expression due to an apparent mutation was previously reported by our group [25]. The current paper provides detailed characterization of this novel rat genetic model and provides strong evidence that titin normally functions to prevent overstretch of the sarcomere.
2. Methods
2.1 Animals and Tissue Sampling
The original rats in which the mutation was observed were from the Sprague-Dawley strain. These animals were crossed with Fisher 344 inbreds. Both original strains were obtained from Harlan Sprague Dawley, Indianapolis, IN. The current homozygote mutant line is 40–50% Fisher 344 and the rest Sprague-Dawley. Animals were maintained on standard rodent chow using protocols approved by the University of Wisconsin-Madison Animal Use and Care Committee. Hearts were removed immediately after euthanasia and the left ventricle, right ventricle, and atria were separated. Samples were obtained from animals ranging from 16 days fetal to 500 days after birth. Tissues for electrophoresis and cDNA analysis were frozen in liquid nitrogen and stored at −80°C.
2.2 Agarose gel electrophoresis and Western blotting
Frozen tissue was dissolved in 8 M urea- 2 M thiourea-3% SDS-75 mM dithiothreitol sample buffer using a small Dounce homogenizer [26] or a MiniBeadBeater (Biospec Products, Bartlesville, OK). Samples were applied to large format SDS agarose gels and the proteins separated electrophoretically. Gels were silver stained and dried between sheets of mylar. Full details of the procedures have been published previously [26].
For Western blotting, protein extracts from wild type and mutant rat left ventricles of 10 day, 49 day and adult (> 1 year) ventricle were dissolved in the presence of urea-thiourea-SDS-dithiothreitol sample buffer (8M urea, 2M thiourea, 50mM Tris/Cl (pH6.8), 5mM dithiothreitol, 3% SDS). The amount of protein was quantified with a BCA protein assay (Pierce, Rockford, IL). Equal amounts of protein from each sample were subjected to large format SDS agarose gels. Proteins were transferred onto a Nitrocellulose membrane (Amersham Biosciences). After blocking, membranes were incubated with antibodies against Novex III [10] (antibody was diluted at 1:200). Subsequently, membranes were incubated with peroxidase-conjugated secondary antibodies (1:2000; Amersham Biosciences) and finally substrate before exposure to CL-XPosure film (Pierce).
2.3 cDNA Cloning and Sequencing
Total RNA was purified from rat ventricle using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA). Samples were obtained from wild type (Wt), heterozygote (Ht), and homozygote (Hm) mutant animals at ages 1, 12, 88, and 500 days. Primers (Supplement Table 1) were designed from nucleotide sequences previously determined for rat cardiac N2BA and rat genomic GenBank Accession Number AF525411 [27] and AC098426.4 respectively. Products were obtained with primers from exon 109 to 225 (includes the full PEVK region) or exon 115 to 225. Exons 111 through 114 were found in all products using the 109 through 225 primers, so these exons were assumed to be present in computing the full PEVK length of each clone in the figures. For PCR amplification the AccuPrime™ Pfx SuperMix system (Invitrogen, California) was used following Invitrogen’s recommendations. PCR products were electrophoretically separated on 0.8% agarose gels containing ethidium bromide. Bands of interest were excised, and the DNA was purified using the Wizard SV gel and PCR clean-up system (Promega, Madison, WI). The blunt ended PCR products were ligated into pGEM®-T Easy vector (Promega, Madison, WI). The ligated DNA was then transformed into E.coli Library Efficiency® DH5α™ Competent Cells (Invitrogen, California) using heat shock. Plasmid DNA was purified using the Wizard Plus SV Minipreps DNA purification system (Promega, Madison, WI) prior to sequencing. In most cases sequences from both strands were obtained. Sequences were identified and compared with Genbank sequences using the NCBI Blast [28], and analyzed with Edit View Version 1.0.1 from ABI Prism™ (Perkin-Elmer Corp., 1996). Exon expression identification was conducted using the ‘Blast 2 sequences’method [29], the rat titin genomic sequence (accession number AC098426) and the human titin genomic sequence (accession number AJ277892 [9]). Rat titin exons were numbered using the homologous numbers from the human sequence. Further details of the methods for RNA extraction, cDNA synthesis, RT-PCR, and sequence analysis have been described in prior publications [26–27].
2.4 Real time RT-PCR analysis
Total RNA was extracted in 1 ml TRIzol (Invitrogen, Frederick, MD), following manufacturer instructions. RNA was precipitated from the TRIzol reagent with isopropanol and re-suspended at 0.5–1.0 mg/ml using RNA secure solution (Ambion, Austin, TX). RNA samples from 1 day, 49 day and adult (1.5 year) rat left ventricle were treated with DNaseI (DNA-free; Ambion, Austin, TX). After extraction, all samples were stored at −80°C. RNA concentration was measured with a NanoDrop ND 1000 spectrometer (NanoDrop Technologies, Wilmington, DE), and RNA integrity was assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). For reverse transcription, 60 ng of RNA were mixed with 5 uM random hexamers, 1 mM each dNTP, 7.5 mM MgCl2, 40 U RNasin (Promega, Madison, WI), 1X PCR buffer II (Applied Biosystems, Foster City, CA) and 250 U of SuperScript II reverse transcriptase (Invitrogen). The reaction was incubated at 25°C for 10 min, 48°C for 45 min, and 95°C for 5 min, then cooled to 4°C. For reverse transcription production, two volumes of ethanol were added with mixing and the mixture incubated at −20°C for 30 minutes. Following centrifugation for 15 minutes at 13,000 rpm and washing with 75% ethanol, the samples were dried. The cDNA was re-suspended in distilled water and used as template. For SYBR Green quantitative real-time PCR, primer pairs (Exon48; Forward; 5’-GACCTCGCAGATGAAGAAG-3’; Reverse; 5’-CTACAGGGAAGGAAAGCAG-3’) were designed for Exon 48 of titin by using Primer Express (Applied Biosystems) according to GenBank Accession No. AC098426. All primer pairs produced a single PCR product as determined by the dissociation curve and gel analysis. Real-time PCR was performed in a 20 ul reaction, 96-well format and 1 X SYBR PCR Master Mix (Applied Biosystems). The reaction was incubated in an Opticon 2 real-time PCR machine (MJ Research) for 40 cycles consisting of denaturation at 95°C for 15 s and annealing/extension at 58–60°C for 1 min. Three samples were measured in each experimental group in quadruplicate, with a minimum of two independent experiments. The relative amount of target mRNA normalized to GAPDH was calculated according to the method described by Pfaffl [30].
2.5 Cardiomyocyte mechanical experiments
Single ventricular myocytes were obtained by enzymatic digestion, using a modification of the technique described previously [12]. Briefly, the heart was rapidly excised from an anesthetized rat and placed in Ca2+-Ringer’s solution. The aorta was cannulated and the heart was placed on a temperature-controlled Langendorff perfusion apparatus. The coronary arteries were perfused with Ca2+-Ringer’s solution for 1–3 minutes to reestablish intrinsic rhythm. The heart was then subsequently perfused with a Ca2+-free Ringer’s for 2–5 minutes, at which time Type II collagenase (0.5mg/ml, Worthington) and hyaluronidase (0.6 mg/ml, Sigma) were added to the perfusate. After ~ 10 minutes, CaCl2 was then added to the perfusate to a final concentration of 0.5 mmol/L and perfusion was allowed to continue for 15 minutes. The heart was removed from the apparatus, and the ventricles were cut into small pieces and placed in a flask containing 10 ml of enzyme solution. The ventricular pieces were gently shaken and subjected to intermittent trituration for 20 minutes. The resulting cellular suspension was filtered and the cells were resuspended in Ca2+-Ringer’s solution containing 200µM Ca2+ and 0.552mg/ml pyruvate. The ventricular myocytes were allowed to settle by gravity and then were rapidly skinned following re-suspension in relaxing solution containing 0.3% Triton X-100 for 10 minutes at room temperature. The skinned ventricular myocytes were washed twice in fresh relaxing solution and stored on ice until use.
Mechanical measurements were performed on an experimental apparatus described previously [31–32]. In brief, approximately 200 µl of a suspension of skinned ventricular myocytes was placed on a glass cover-slip. Ventricular myocytes were attached with the use of silicone adhesive (Aquarium Sealant, Dow Corning) to the tips of steel pins (outer diameter, 10 µm), which were in turn fixed to a force transducer (model 400A, Aurora Scientific) and motor (model 308, Aurora Scientific). After attachment, the silicone adhesive was allowed to cure for 30–45 minutes. The output signal from the force transducer was amplified 10-fold. Signals representing force and motor position (equivalent to myocyte length) were sampled and saved to computer files using SLControl Software [33]. Sarcomere length and cell width were monitored throughout the duration of the experiment by video imaging.
At the beginning of each experiment, the preparation was shortened to slack length and the cross-sectional area of the myocyte was determined (assuming a circular profile) using video microscopy. The length-tension relationship, previously described [12], for the preparation was then assessed from a series of trials during each of which the myocyte was first shortened to slack length for 500 ms and then extended by increasing percentages of fiber length. Resting tension was measured before the stretch and after steady state had been achieved following the stretch (~18.5 s). Individual trials were performed every 1–2 minutes. The interval between cycles was sufficient to ensure that resting tension had reached a steady state between measurements.
2.6 Statistical analysis
All values are expressed as means ± SE. Differences between groups were evaluated using the Student’s t-test with probabilities of 95% or greater considered statistically significant.
3. Results
3.1 Genetics and isoform patterns
Our previous studies demonstrated that cardiac titin undergoes a series of isoform transitions during development as observed by SDS-agarose electrophoresis [12]. A few rats from this study, however, had ventricle titin patterns inconsistent with their ages, and their profiles appeared similar to those from much younger animals [25]. All the animals with the altered profiles were from the same litter, and this immediately suggested there was a genetic factor involved.
The 7 animals from litter 2 used in the original age study [12] are shown at the top left in the pedigree (Figure 1). The mother of the litter had been sacrificed for the original age study, so the father must have been the first carrier. Attempts to identify additional carriers from the Harlan source were unsuccessful [25]. A brother-sister (number 38) mating resulted in three offspring that all had the altered titin pattern. Mating the father and mother separately to Fisher 344 rats showed that only the mother (38) was a carrier. Subsequently, two other litter 2 males (numbers 35 and 29) were found to transmit the altered titin pattern to their offspring when crossed with wild type females.
Figure 1.

Pedigree for the rat mutation that alters titin alternative splicing.
A littermate from the original set of affected animals, when mated to a wild type Fisher 344, yielded offspring with two types of titin agarose gel patterns (Figure 2A): (1) the wild type, with approximately equal proportions of N2B (the major adult form), N2BA-N1 (similar in size to human soleus titin −3.7 MDa), and N2BA-N2 bands (the latter two present in late embryonic and early post-natal ventricles) and (2) the altered pattern, where the primary band had a migration position equal to or slightly slower than the N2BA-N1. The N2B band in the latter animals was totally absent. Both males and females were found with the altered titin isoforms.
Figure 2.
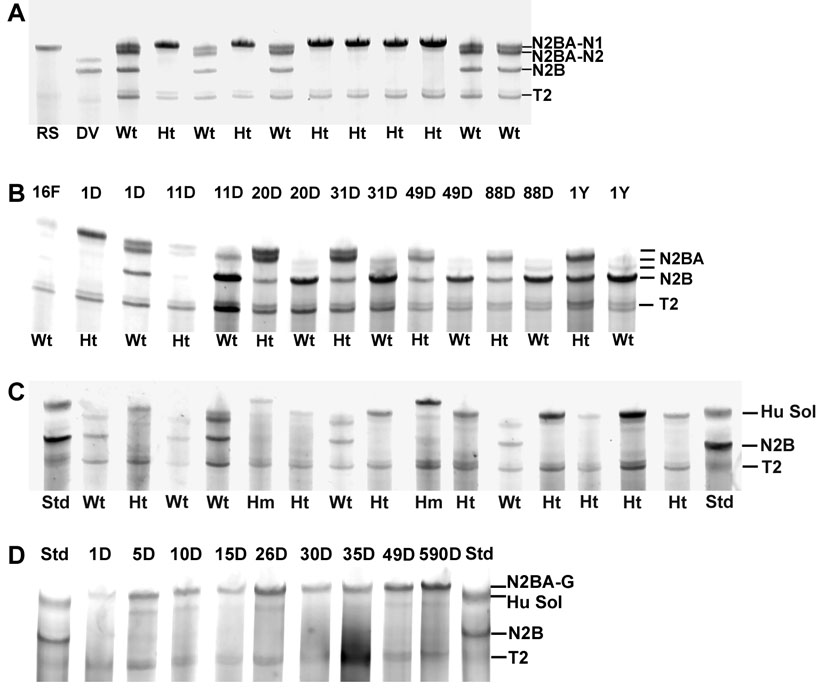
SDS agarose gels of rat ventricle. A. Samples from a one day old litter of rat pups. Wt – wild type; Ht – heterozygotes. Wild type animals have three major titin bands in addition to the T2, a titin proteolytic degradation band. Heterozygotes have primarily a single band equivalent to or slightly larger than the large neonatal titin (N2BA-N1). RS – rat soleus; DV – dog ventricle. B. Age comparisons of wild type and heterozygotes. All the samples were run on the same gel except the one year wild type. 16F – sixteen day fetal. C. Off spring (12 days of age) from a cross of two heterozygotes. Three different phenotypes were present – the wild type and heterozygote patterns as in (A) and a new larger sized, slower migrating band that came from potential homozygotes (Hm). Standards contained human soleus (Hu Sol – 3.7 MDa) and adult rat soleus (primarily N2B – 3.0 MDa). D. Developmental patterns of titin isoforms in homozygotes from 1 to 590 days of age.
The previous results showed that small amounts of the N2B isoform gradually appeared in older affected animals so that it constituted about 20% (in contrast to 80–85% in wild type rats) of the total intact titin at 25 days of age (the last time point mutant observed previously) [25]. Comparisons between heterozygote mutants and wild type animals have now been extended to one year of age (Figure 2B). Whereas the titin isoform transition in wild type animals was complete by 20–25 days of age, changes are still occurring much later in affected rats. Even at one year of age, the full transition to the wild type pattern did not occur, and the titin is approximately 50% N2B and 50% of a larger species that migrates in the same position as the N2BA-N1 isoform (see [25]). This pattern remained stable in hearts from animals aged greater than two years (data not shown).
Crossing two affected animals (Figure 1, animal numbers 203 and 204, presumably heterozygotes) resulted in offspring with three titin isoform phenotypes in hearts from early postnatal rats (Figure 2C). Both the early wild type pattern with three titin gel bands and the heterozygotes with a single major band (N2BA-N1) were observed. However, some rats had a new pattern with a major band with apparent size of 3.9 MDa that we have labeled N2BA-G (for giant). This is the largest vertebrate titin isoform described to date and it was assumed to occur in animals homozygous for the mutation.
Crossing animal 38 with another heterozygote carrier (167) resulted in a homozygote carrier (270, verified by a separate crossing with a wild type that resulted in all heterozygote offspring) (Figure 1). Matings between homozygous carriers (ie. 509 and 431) resulted in 100% homozygous offspring with similar giant sized titins (Figure 2D). Homozygotes, in contrast to heterozygotes, showed no evidence of transition to smaller isoforms, even through 590 days of age. The gel patterns show a small amount of a band between 3.6 and 3.7 MDa (most visible in the animals at 5 and 10 days of age). It is not clear whether this is a trace of a shorter isoform or a proteolytic degradation product of the giant band. All the progeny patterns and resultant titin phenotypes are consistent with a Mendelian inheritance and an autosomal dominant type mutation.
3.2 heart size
In light of the dramatic difference in titin size between wild type and homozygous mutants, it might be expected that some type of cardiac abnormality might occur. Weights of left ventricle, right ventricle, and atria are shown in Figure 3. The sizes of the heterozygote and homozygote mutant left ventricles were somewhat greater, but the changes were not different statistically. The size of the atria, however, was significantly (P<0.05) larger in the homozygous mutants. This suggests that the atria were undergoing additional stress in the mutants.
Figure 3.

Sizes of different regions of the heart between wild type (Wt), heterozygotes (Ht), and homozygote mutants (Hm). The left ventricle weight includes both the septum and the free wall. Only the atrial sizes were significantly different between the homozygous mutants and the other genetic groups. Wt, N=6; Ht, N=7; Hm, N=6.
3.2 Does the mutation affect another muscle alternatively spliced protein?
The next question we addressed was whether the apparent mutation affected alternative splicing in any other myofibrillar proteins besides titin. Cardiac troponin T has been shown previously to both undergo developmental transitions and be alternatively spliced [34–35], so wild type and homozygous mutant rats were examined at ages between 1 and 31 days (Figure 4). Western blots from wild type animals at one day of age showed a major upper embryonic-neonatal band and a lower band with less staining intensity. By 10 days of age and thereafter this lower band became dominant. Samples from homozygous mutant animals (Figure 4 lower panel) showed a similar pattern of isoform transition as occurred in the wild type. Thus there appeared to be no significant effect on the troponin T developmental time course in the titin mutant animals. Whatever is altering the titin splicing pattern does not appear to have dramatic effects on the developmental changes in troponin T isoforms.
Figure 4.

Western blots with an antibody against troponin T of wild type (Wt) and homozygote mutant (Hm) ventricle from animals 1 to 31 days of age. The upper fetal type is gradually replaced by the lower adult form. The transition time course is similar in both groups of rats.
3.3 PEVK exon expression patterns at various ages in wild type and mutant rats
The extra apparent mass of the N2BA titin isoforms in homozygote mutants suggested that there may be additional exons expressed. In particular focus was placed on the PEVK region where previous work had shown that more PEVK exons were expressed in wild type embryonic ventricle than in the post-natal tissue [22]. It was also observed that the number of PEVK exons expressed in fetal rat heart was still significantly fewer than that found previously in human soleus titin [22]. RT-PCR was performed with primers between exons 115 and 225 using wild type, heterozygote, and homozygote mutant animals at 1, 12, 88, and 500 days of age. A minimum of 5 clones were sequenced for each age and mutation status. The average number of amino acids for each PEVK clone at the different ages is shown in Figure 5. Exon patterns for each of the PCR clones are presented in Supplemental Figure S1, S2, S3, and S4. Heterogeneity in exon patterns in different clones from wild type ventricles was observed at each of the different ages in agreement with previous studies [22]. Variation between clone sequences within an age group was somewhat reduced in the homozygote mutants. Only the 500 day homozygote clones were all identical. PEVK amino acid number declined with age for all three mutation-status groups. Significant differences (P<0.01) using T-test comparisons between pairs of wild type (Wt), heterozygote (Ht), and homozygote (Hm) mutants were observed at ages 1 and 12 days, with the numbers being Ht > Hm > Wt. However, the PEVK size did not vary significantly between groups (P= 0.16–0.86) in hearts at 88 or 500 days of age.
Figure 5.

Comparisons of amino acid length of the PEVK in wild type, heterozygotes, and homozygotes at various ages. Error bars depict standard deviations. Differences between mutation status groups are not statistically significant at 88 or 500 days of age.
3.4 Novex III in wild type and homozygous mutants
Novex III is a large exon found in the titin gene preceding the N2B unique position [10]. A shorter titin isoform (~700 KDa) has also been found that contains amino acids coded by this exon at the carboxyl terminal end. The protein sequence from this exon contains several Ig domains plus additional unique sequence. The possibility that the Novex III exon coded for part of the giant titin from homozygotes was explored by Western blots and quantitative PCR. Western blots from agarose gels gave a single reactive band in the ~700 KDa region (Figure 6). There was no evidence of reaction with any of the full length titin bands from 10 day, 49 day, or adult wild type or homozygote samples. Thus the Novex III does not appear to be a part of the giant titin.
Figure 6.

Western blot with antibody against Novex III. Wild type and homozygote samples were prepared from animals at 10 day, 49 day, and adult (> 1 year) of age. Left – Western blot; right – silver stained companion gel with the same sample lanes and protein loads.
Quantitative PCR was conducted to determine the amount of message containing the Novex III exon. The results showed no difference between the wild type and homozygote mutants at ages 10 or 49 days, but there appeared to be a modest increase among homozygous adult samples (approximately 1.7–1.9 compared to wild type with three biological replicates)(Figure S5).
3.5 Passive tension comparisons between wild type and homozygous mutants
Titin has been postulated to be the major contributor to passive tension within the physiological working range of sarcomere lengths in the heart [33]. If this is the case, then it might be expected that there would be large passive tension differences between wild type and homozygous mutant myocytes since the predominant titin size is 3.0 MDa and 3.9MDa in the respective hearts. Individual cardiomyocytes were obtained by digestion and subjected to a stretch protocol. Mean rest tension measurements were plotted against sarcomere length (Figure 7). Cells were stretched in 2.5% increments with slackening back to the rest stage after each stretch. Passive tension of wild type cells rose rapidly at sarcomere lengths between 2.0 and 2.5 micrometers. Cell attachments broke at an average sarcomere length of 2.48 ± 0.05µm with wild type myocytes (Table 1). In contrast, the homozygous mutant cells could be stretched by more than 100% without breakage (Figure 7). The developed rest tension was nearly twice as great at 4 µm as that of the wild type cells at the point of rupture. Stretching cells by 25% led to an obscuring and distortion of the sarcomere appearance in the wild type cells, but clear banding patterns were still observed in homozygous mutant cells stretched the same percentage (Figure 8). Homozygote mutant myocytes were significantly longer and wider than their wild type counterparts (homozygotes L = 106 ± 3 µm, W = 67 ± 4 µm)(wild type L = 64 ± 2 µm, W = 28 ± 2 µm). Slack sarcomere lengths were also 0.14 µm longer in mutant cells compared to wild type (Table 1).
Figure 7.

Resting tension comparisons between single enzymatically isolated cardiomyocytes from wild type and homozygote mutant ventricles. Note that homozygote mutant cells could be stretched to at least 4 µm without breakage.
Table 1.
Cardiomyocyte mechanics
| Significance | Wild Type | Homozygote Mutant | |
|---|---|---|---|
| Myocyte length* (µm) | 64 ± 2 | 106 ± 3 | P< 0.001 |
| Myocyte width* (µm) | 28 ± 2 | 67 ± 4 | P<0.001 |
| Slack sarcomere length (µm) | 1.88 ± 0.04 | 2.02 ± 0.04 | P<0.01 |
| Mean failure sarcomere length (µm) | 2.48 ± 0.05 | No failure to SL > 4.0 µm | |
| Mean percent stretch at failure | 32 ± 2 | No failure at 100% stretch | |
| Mean failure force (N/m2) | 2.66 ± 0.32 | >5.18 ± 0.74 (last recorded force) | |
Myocyte length and width measurements were obtained from images of bulk digests.
Figure 8.

Light micrograph patterns of wild type (left pair) and homozygous mutant (right pair). Myocytes. Cells were stretched by 5% above their passive length (upper) or by 25% (lower). While the sarcomere patterns become significantly obscured by stretch of 25% to a sarcomere length of 2.25 µm in wild type cells, sarcomere patterns were still prominent when stretched to 2.7 µm in the homozygote mutants.
4. Discussion
The current paper describes a novel mutation that dramatically affects the titin isoform expression patterns in the rat heart. All the evidence to date indicates that the mutation follows Mendelian inheritance with autosomal dominance. The altered pattern has been transmitted through 7 generations to date. Rats carrying the mutation show no external phenotype, and even homozygous carriers live and reproduce. The lack of apparent major health effects is rather surprising since the titin isoform pattern is approximately 85% of the 3.0 MDa N2B isoform in wild type animals [12] while the homozygote mutants have greater than 90% of a giant isoform with a size near 3.9 MDa. Thus the giant mutant protein is nearly one third larger than the native protein. Since titin is linked both to the Z line and to the M line in each half sarcomere, the fact that the heart remains functional demonstrates the remarkable flexibility of this unusual protein. Several genes that are altered in heart failure (for example, Atrial naturetic factor [ANF] and β-myosin heavy chain) are also up-regulated in the homozygous mutants as determined using microarrays and quantitative PCR; supporting data will be presented in a separate full report.
The significantly larger size of the giant titin in the homozygous mutants led us to postulate that protein sequence from additional exons must be included. Previous studies with human and rat titin cDNA have outlined the splicing patterns for several of the major isoforms [8–10, 12, 25, 27]. Most of the titin splicing occurs in the PEVK region of the I band and in exons coding for a series of Ig domains that are on the Z line side of the PEVK (the so called middle Ig region; see Figure 1 [27]). The largest isoform described to date is that from human soleus with a size of approximately 3.7 MDa [8]. This isoform contains almost all the exons for the middle Ig region and the majority of the exons coding for the PEVK region (giving a total of more than 2000 amino acids with >200,000 Daltons of mass [8]). It does not contain the N2B unique region, coded by exon 49 that is only expressed in cardiac muscle. Evidence from cDNA sequencing has been presented previously that the 3.7 MDa rat embryonic titin isoform (referred to as N2BA-N1 [12]) contains all of the middle Ig domains but a somewhat shorter PEVK than found in human soleus. So our initial hypothesis was that the giant titin contained the full middle Ig sequence plus sequence from most or all of the PEVK exons. Statistically significant differences in the number of PEVK amino acid between wild type, heterozygotes, and homozygotes in hearts from rats aged one and 12 days were observed, but the heterozygotes were longer than the homozygotes and wild type. The length of the PEVK declined with age in all groups; however, the maximum difference between wild type and homozygote mutants was only about 17,000 Daltons out of the total titin size of 3.0–3.9 MDa. In addition, the PEVK length was much shorter (less than half) of that for human soleus in all PCR clones obtained. Thus our original hypothesis of a longer PEVK region in the homozygotes proved to be incorrect and could not be used to explain the additional size of the giant titin. Similarly there was no evidence that part of the increased size was due to the inclusion of sequence from the large Novex III exon (Figure 6). Post-translational modifications (i.e. phosphorylation, glycosylation, etc.) may alter protein migrations and give apparent changes in molecular weight. Changes in phosphorylation seem unlikely since most of the protein chain is identical between the wild type N2BA isoforms and the giant titin. Large changes in glycosylation appear even less rational since such extensive changes rarely occur in intracellular proteins.
Our previous studies have shown that the alternatively splicing patterns in the PEVK region of the N2BA isoforms were extremely heterogeneous [12, 25, 27]. Ten separate clones from a single PCR reaction using human cDNA were found to have different alternatively spliced patterns [25]. It was anticipated that the mutation affecting titin isoform expression might have reduced heterogeneity in exon splicing patterns. Comparisons of the clones obtained from the different phenotypic groups showed this to be true for the day 1 and day 500 homozygote samples where 3 of the 4 clones had identical sequences and 5 of the 5 were the same in these respective groups (Figure S1, S4). However, the day 12 homozygotes had only a pair of the 12 clones that were identical and the day 88 homozygotes had 3 different clone sequences among the 5 total examined (Figure S2, S3). Thus, the heterogeneity of PEVK sequences was reduced but not eliminated in the mutants.
Although most of the developmental changes in protein isoforms in the myofibril involve changes in genes expressed (i.e. cardiac β to α myosin heavy chain, slow skeletal troponin I to cardiac troponin I, etc), troponin T is a notable exception since there is a single cardiac gene and different alternatively spliced isoforms. The cardiac troponin T is expressed as a longer protein form at the embryonic stage but is subsequently replaced by a shorter isoform resulting from transcripts with fewer exons [34–35]. If the mutation affecting titin alternative splicing affects global splicing, we would expect that the troponin T might show persistence of the embryonic form into adulthood. However, Western blots demonstrated that essentially normal transitions and time courses were found with wild type and homozygous mutants when compared at ages between 1 and 31 days (Figure 4). Thus the mutation did not appear to be affecting all alternatively spliced proteins.
Studies have demonstrated that the passive tension of skeletal muscle cells is related to the size of the titin [36–39]. In cardiac muscle such tension correlates with the relative ratio of the longer N2BA isoform species to that of the shorter N2B [11, 40]. It was anticipated that myocytes from the homozygous mutants, with their giant sized titin, would have reduced passive tension compared to wild type, and this was confirmed (Figure 7). While rest tension rose rapidly with sarcomere length extension in wild type myocytes, the tension rose more gradually in the homozygous mutants and the cells could be extended to lengths greater than 4 microns. Wild type cells broke upon stretching approximately 32% (an average sarcomere length of 2.48 µm), but the mutant cells did not pull loose even when stretched to double their resting length (100% stretch). In addition the total tension per cross sectional area in the mutant cells was nearly double compared to the point of failure in the wild type myocytes. The average cell size was significantly larger in the homozygous mutants than wild type (Table 1). However, the cells actually used for the rest tension measurements were more closely matched in diameter (W = 21 ± 1 µm in wild type and 28 ± 2 µm in homozygous mutants). Neither rest tension per cross sectional area or failure sarcomere length were correlated with fiber cross-sectional area (data not shown).
It is unclear what is the source of the additional passive tension at very long sarcomere lengths in the mutants (mutants have nearly double the rest tension at 4 µm sarcomere length than wild type cells at failure). The shape of the force versus sarcomere length curve with the mutant cells was also more linear than would have been expected from previous studies with cells containing titins of various lengths. Possibilities to explain these differences include the intracellular intermediate filament network or forces arising from the remaining sarcolemma. The collagen extracellular matrix should have been removed by collagenase during the cell preparation. Since the cells are stretched beyond the thick and thin filament overlap distance, it also appears unlikely that residual cross-bridge forces are involved.
Titin and associated proteins have been proposed to function as a stretch sensor to modify protein expression in the heart [41–42]. If the homozygous mutant hearts operate in the same sarcomere length range as typical wild type cells (~1.8 to 2.2 µm), then any stretch signal from the titin ought to be reduced or abolished. The current rat model should prove to be a useful system to investigate titin’s role in signal transduction.
Supplementary Material
Acknowledgments
This work was supported by the College of Agricultural and Life Sciences, University of Wisconsin-Madison and National Institutes of Health grants RO1 HL77196 (M.L.G) and R37 HL82900 (R.L.M.). The gift of the Novex-III antibody from Siegfried Labeit is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang K, McClure J, Tu A. Titin: major myofibrillar components of striated muscle. Proc Natl Acad Sci USA. 1979;76:3698–3702. doi: 10.1073/pnas.76.8.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maruyama K. Connectin, an elastic protein from myofibrils. J Biochem (Tokyo) 1976;80:405–407. doi: 10.1093/oxfordjournals.jbchem.a131291. [DOI] [PubMed] [Google Scholar]
- 3.Granzier HL, Labeit S. Titin and its associated proteins: the third myofilament system of the sarcomere. Adv Protein Chem. 2005;71:89–119. doi: 10.1016/S0065-3233(04)71003-7. [DOI] [PubMed] [Google Scholar]
- 4.Granzier H, Wu Y, Siegfried L, LeWinter M. Titin: physiological function and role in cardiomyopathy and failure. Heart Fail Rev. 2005;10:211–223. doi: 10.1007/s10741-005-5251-7. [DOI] [PubMed] [Google Scholar]
- 5.LeWinter MM, Wu Y, Labeit S, Granzier H. Cardiac titin: structure, functions and role in disease. Clin Chim Acta. 2007;375:1–9. doi: 10.1016/j.cca.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 6.Lange S, Ehler E, Gautel M. From A to Z and back? Multicompartment proteins in the sarcomere. Trends Cell Biol. 2006;16:11–18. doi: 10.1016/j.tcb.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Linke WA. Sense and stretchability: The role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2007 doi: 10.1016/j.cardiores.2007.03.029. in press. [DOI] [PubMed] [Google Scholar]
- 8.Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 9.Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res. 2000;86:1114–1121. doi: 10.1161/01.res.86.11.1114. [DOI] [PubMed] [Google Scholar]
- 10.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 11.Cazorla O, Freiburg A, Helmes M, Centner T, McNabb M, Wu Y, et al. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res. 2000;86:59–67. doi: 10.1161/01.res.86.1.59. [DOI] [PubMed] [Google Scholar]
- 12.Warren CM, Krzesinski PR, Campbell KS, Moss RL, Greaser ML. Titin isoform changes in rat myocardium during development. Mech Dev. 2004;121:1301–1312. doi: 10.1016/j.mod.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res. 2004;94:967–975. doi: 10.1161/01.RES.0000124301.48193.E1. [DOI] [PubMed] [Google Scholar]
- 14.Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94:505–513. doi: 10.1161/01.RES.0000115522.52554.86. [DOI] [PubMed] [Google Scholar]
- 15.Kruger M, Kohl T, Linke WA. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-I isoform switching are not critically triggered by birth. Am J Physiol Heart Circ Physiol. 2006;291:H496–H506. doi: 10.1152/ajpheart.00114.2006. [DOI] [PubMed] [Google Scholar]
- 16.Warren CM, Jordan MC, Roos KP, Krzesinski PR, Greaser ML. Titin isoform expression in normal and hypertensive myocardium. Cardiovasc Res. 2003;59:86–94. doi: 10.1016/s0008-6363(03)00328-6. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, et al. Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation. 2002;106:1384–1389. doi: 10.1161/01.cir.0000029804.61510.02. [DOI] [PubMed] [Google Scholar]
- 18.Rankinen T, Rice T, Boudreau A, Leon AS, Skinner JS, Wilmore JH, et al. Titin is a candidate gene for stroke volume response to endurance training: the HERITAGE Family Study. Physiol Genomics. 2003;15:27–33. doi: 10.1152/physiolgenomics.00147.2002. [DOI] [PubMed] [Google Scholar]
- 19.Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, et al. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.cir.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 20.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, et al. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. [DOI] [PubMed] [Google Scholar]
- 21.Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, et al. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708–716. doi: 10.1161/01.RES.0000143901.37063.2f. [DOI] [PubMed] [Google Scholar]
- 22.van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, et al. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 23.LeWinter MM. Titin isoforms in heart failure. Are there benefits to supersizing? Circulation. 2004;110:109–111. doi: 10.1161/01.CIR.0000137284.17083.93. [DOI] [PubMed] [Google Scholar]
- 24.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2004;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 25.Greaser ML, Krzesinski PR, Warren CM, Kirkpatrick B, Campbell KS, Moss RL. Developmental changes in rat cardiac titin/connectin: transitions in normal animals and in mutants with a delayed pattern of isoform transition. J Muscle Res & Cell Motil. 2005;26:325–332. doi: 10.1007/s10974-005-9039-0. [DOI] [PubMed] [Google Scholar]
- 26.Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high molecular weight proteins. Electrophoresis. 2003;24:1695–1702. doi: 10.1002/elps.200305392. [DOI] [PubMed] [Google Scholar]
- 27.Greaser ML, Berri M, Warren CM, Mozdziak PE. Species variations in cDNA sequence and exon splicing patterns in the extensible I-band region of cardiac titin: relation to passive tension. J Muscle Res & Cell Motil. 2002;23:471–480. doi: 10.1023/a:1023410523184. [DOI] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 30.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strang KT, Sweitzer NK, Greaser ML, Moss RL. β-adrenergic receptor stimulation increases unloaded shortening velocity of skinned single ventricular myocytes from rats. Circ Res. 1994;74:542–549. doi: 10.1161/01.res.74.3.542. [DOI] [PubMed] [Google Scholar]
- 32.Fitzsimons DP, Moss RL. Strong binding of myosin modulates length-dependent Ca2+ activation of rat ventricular myocytes. Circ Res. 1998;83:602–607. doi: 10.1161/01.res.83.6.602. [DOI] [PubMed] [Google Scholar]
- 33.Campbell KS, Moss RL. SLControl: PC-based data acquisition and analysis for muscle mechanics. Am J Physiol Heart Circ Physiol. 2003;285:H2857–H2864. doi: 10.1152/ajpheart.00295.2003. [DOI] [PubMed] [Google Scholar]
- 34.Saggin L, Ausoni S, Gorza L, Sartore S, Schiaffino S. Troponin T switching in the developing rat heart. J Biol Chem. 1988;263:18488–18492. [PubMed] [Google Scholar]
- 35.Jin JP, Huang QQ, Yeh HI, Lin JJ. Complete nucleotide sequence and structural organization of rat cardiac troponin T gene. A single gene generates embryonic and adult isoforms via developmentally regulated alternative splicing. J Mol Biol. 1992;227:1269–1276. doi: 10.1016/0022-2836(92)90540-z. [DOI] [PubMed] [Google Scholar]
- 36.Horowits R. Passive force generation and titin isoforms in mammalian skeletal muscle. Biophys J. 1991;61:392–398. doi: 10.1016/S0006-3495(92)81845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K, McCarter R , Wright J, Beverly J, Ramirez-Mitchell R. Regulation of skeletal muscle stiffness and elasticity by titin isoforms: a test of the segmental extension model of resting tension. Proc Natl Acad Sci USA. 1991;88:7101–7105. doi: 10.1073/pnas.88.16.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–1044. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neagoe C, Opitz CA, Makarenko I, Linke WA. Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil. 2003;24:175–189. doi: 10.1023/a:1026053530766. [DOI] [PubMed] [Google Scholar]
- 40.Fukuda N, Wu Y, Nair P, Granzier HL. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J Gen Physiol. 2005;125:257–271. doi: 10.1085/jgp.200409177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lange S, Ehler E, Gautel M. From A to Z and back? Multicompartment proteins in the sarcomere. Trends Cell Biol. 2006;16:11–18. doi: 10.1016/j.tcb.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Hoshijima M. Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am J Physiol Heart Circ Physiol. 2006;290:H1313–H1325. doi: 10.1152/ajpheart.00816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.