

Abstract
An efficient, stereospecific synthesis of the alkaloids senepodine G (2) and cermizine C (1) has been completed using the BF3·Et2O promoted stereospecific addition of Me2CuLi toα,β-unsaturated lactam 6 to provide lactam 3, the addition of MeMgBr followed by HCl to convert 3 to senepodine G (2) (6 steps, 40% overall yield) and the stereospecific NaBH4 reduction of 2 to give cermizine C (1) (7 steps, 40% overall yield).
Kobayashi recently reported the isolation of the lycopodium alkaloid cermizine C (1) from the club moss Lycopodium cernuum and the related alkaloid senepodine G (2) from the club moss Lycopodium chinense (see Scheme 1).1,2,3 The structures were elucidated by 1D and 2D NMR spectroscopic methods. Senepodine G (2) is cytotoxic to murine lymphoma L1210 cells with an IC50 of 7.8 μg/mL. The absolute configuration of other co-occurring more complex natural products were assigned by modified Mosher’s method suggesting that the absolute configurations of 1 and 2 are as drawn.
SCHEME 1.

Retrosynthesis of Cermizine C (1) and Senepodine G (2)
We set out to synthesize these novel alkaloids as a means of developing chemistry that might be useful for more complex members of these families. We thought that it should be possible to prepare cermizine C (1) from senepodine G (2) by reduction of the iminium cation with NaBH4. Although similar reductions are known,2b,4 the stereoselectivity of this reduction is uncertain because steric interactions with the methyl group should direct hydride attack from the bottom face, whereas steric interactions with the right hand ring should direction hydride attack from the top face. Iminium cation 2 can be prepared by addition of MeMgBr to lactam 3 followed by treatment with HCl. We conceived of two approaches to lactam 3. We hoped that it might be possible to carry out a directed conjugate reduction of unsaturated Meldrum’s acid derivative 4 to give 5, which would cyclize to form lactam 3. Alternatively, it might be possible to add methylcuprate stereospecifically to unsaturated lactam 6.
Knoevenagel condensations between a ketone and a β-dicarbonyl compound are typically carried out using an ammonium acetate catalyst. Since (±)-pelletierine (7)5 contains a secondary amine we treated it with 1 equiv of AcOH to form the acetate salt. Heating this salt with 2.4 equiv of Meldrum’s acid in EtOH required 2 days at 60 °C for the complete consumption of the pelletierine. To our surprise, we did not isolate 4, but rather the β,γ-unsaturated lactam 11 in 68% yield. Equilibration of 11 with K2CO3 in MeOH for 1 day afforded a 3:1 mixture of α,β-unsaturated lactam 13 and β,γ, -unsaturated lactam 11. Treatment of pure 13 afforded the identical 3:1 mixture indicating that equilibrium had been reached. Although α,β-unsaturated carbonyl compounds are usually much more stable than their β,γ, -unsaturated isomer, there are examples of unsaturated amides in which significant amounts of both isomers are present at equilibrium.6 Ban prepared 13 by treatment of N-acetylpelletierine with triethyloxonium tetrafluoroborate in CH2Cl2 and then treatment of the resulting intermediate with t-BuOK in t-BuOH.7 These conditions presumably led to the same 3:1 equilibrium mixture of 13 and 11 that we observed.
Presumably the Knoevenagel condensation of 7 and Meldrum’s acid occurred to give 4, which cyclized readily to give the conjugated lactam acid 8. Equilibration afforded the unconjugated isomer 9, which decarboxylated to give enol 10. Kinetic protonation of 10 on the α-carbon formed the β,γunsaturated lactam 11. These reaction conditions, with the amine neutralized as the acetate salt, are mild enough to prevent equilibration to give α,β-unsaturated lactam 13.
Hydrogenation of either 11 or the 3:1 mixture of 11 and 13 over PtO2 in EtOH at 50 psi H2 for 6 h as described by Ban afforded a 16:1 mixture of lactams 12 and 3 in 96% yield. Addition of MeMgBr (4 equiv) in THF to 12 at 60 °C for 2.5 h followed by addition of 3 M HCl in MeOH afforded (±)-7-epi-senepodine G (14) in 100% yield. A similar reaction at 25 °C gave 14 in 76% yield. Reduction of 14 with NaBH4 in MeOH at 25 °C for 10 min occurred stereospecifically by axial attack from the less hindered top face to provide (±)-5-epi-cermizine C (15) in 82% yield.8 The stereochemistry of 15 was established by NOE studies (see supporting material) permitting the assignment of the stereochemistry of the major lactam as 12, not 3.
Since we could not isolate 4 and explore the directed conjugate reduction to give 5, we turned our attention to the alternate route to senepodine G starting with unsaturated lactam 6. Batey prepared amide alcohol 17 by protection of 2-piperidineethanol (16) with TBDMSCl, acylation, and deprotection.9 We found that reaction of (S)-1610 with 3 equiv of (diethoxyphosphoryl)acetyl chloride11 and 3.1 equiv of Et(i-Pr2)N in THF afforded the crude ester amide. The ester was selectively hydrolyzed with K2CO3 in EtOH at 60 °C for 4.5 h to provide amide alcohol 17 in 81% yield (see Scheme 3). Dess-Martin oxidation (79% yield) and intramolecular Horner-Wadsworth-Emmons Wittig reaction (80% yield) as described by Batey9 provided a practical route to optically pure α,β-unsaturated lactam (S)-6.
SCHEME 3.
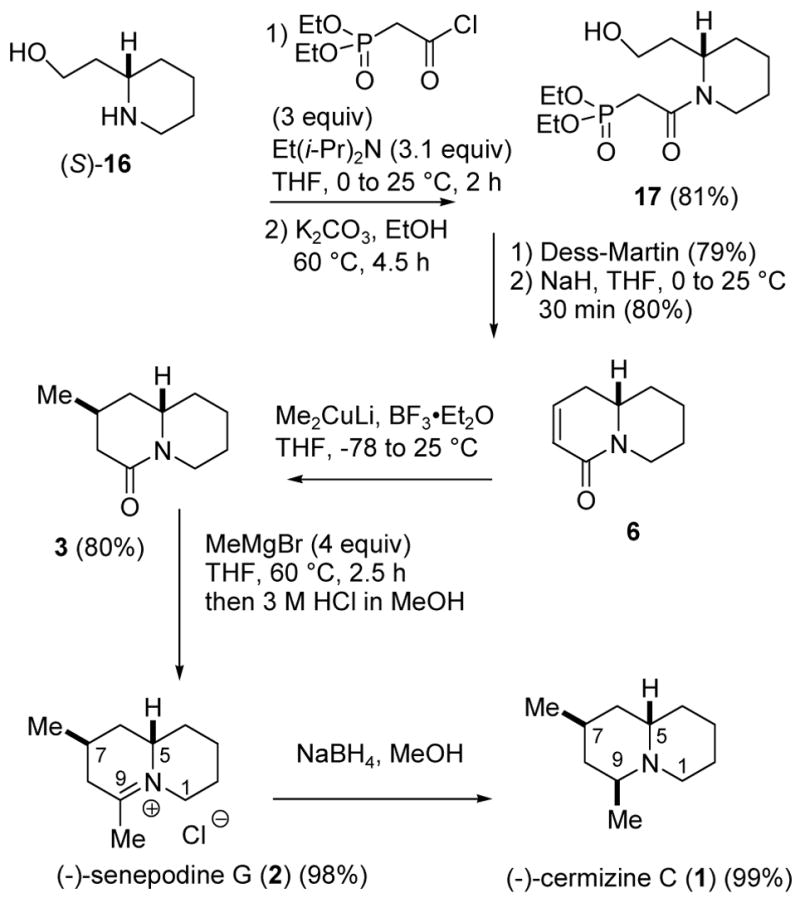
Synthesis of Cermizine C (1) and Senepodine G (2)
Conjugate addition of Me2CuLi promoted by BF3·Et2O12 occurred selectively by axial attack from the less hindered top face to give a >99:1 mixture of the desired lactam 3 and the epimeric lactam 12 in 80% yield. A similar addition of Me2CuLi promoted by TMSCl13 gave a 14:1 mixture of 3 and 12 in 77% yield. Addition of MeMgBr (4 equiv) to 3 in THF at 60 °C for 2.5 h followed by addition of 3 M HCl in MeOH afforded a 98% yield of (−)-senepodine G (2), with spectral data identical to those reported for the natural product.1 Lactam 3 was recovered unchanged from treatment with MeMgBr at 25 °C, although the epimeric lactam 12 was converted to 14 in 76% yield under these conditions. Presumably, the methyl group blocks the convex face of 3. The methyl group of 12 is on the concave face making the top, convex face less hindered so that addition of MeMgBr occurs under milder conditions.
Reduction of 2 with NaBH4 in MeOH at 25 °C for 10 min provided (−)-cermizine C (1), which was isolated as its hydrochloride salt in 82% yield.8 The spectral data for cermizine hydrochloride (1) are identical to those reported for the natural product,1 which was isolated in minute amounts (2.2 mg) and not indicated to be the protonated form. Hydride addition to 2 took place stereospecifically by axial attack from the bottom face. Apparently the directing effect of the methyl group is greater than that of the right hand ring (see Figure 1).
Figure 1.
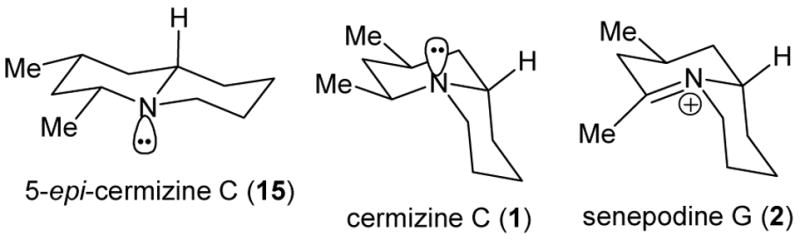
Stable conformations of 5-epi-cermizine C (15), cermizine C (1) and senepodine G (2).
Neutralization of the hydrochloride salt of 1 gave the free amine, which is not stable in air. On the other hand, the free base of 5-epi-cermizine C (15) is stable in air. The stable conformation of 5-epi-cermizine C (15) is a trans-quinolizidine, as indicated by a Bohlmann band at 2783 cm−1, while the most stable conformation of cermizine C (1) was shown to be a cis-quinolizidine by Kobayashi (see Figure 1).1 cis-Quinolizidines have been reported to react with air to give N-oxides more readily than trans-quinolizidines.14 Quinolizidines are also known to readily scavenge carbon dioxide from the air to form bicarbonate salts.15
The optical rotation of synthetic senepodine G (2), [α]D −77, has the same sign as that of natural senepodine G, [α]D −35,1 suggesting that the natural product has the absolute configuration shown. The difference in magnitude may result from errors resulting from the small amount (1 mg) of the natural product available. The optical rotation of synthetic cermizine C (1), [α]D −2, has the opposite sign as that of natural cermizine C, [α]D +4.1 However, Kobayashi indicated that the [α]D value of cermizine C is not reliable due to the small amount of sample and small value of the optical rotation. It is most likely that cermizine C has the same absolute configuration as senepodine G, although this need not be the case since they were isolated from different Lycopodium species. Because of the low value, the [α]D of cermizine C is not very useful. We have therefore provided the CD spectra of synthetic senepodine G (2) and cermizine C (1) from 200–400 nm in the Supporting Information These data may be more useful for comparison purposes.
In conclusion, we have developed an efficient, stereospecific synthesis of the alkaloids senepodine G (2) and cermizine C (1) using the BF3·Et2O promoted stereospecific addition of Me2CuLi to α,β-unsaturated lactam 6 to give lactam 3, the addition of MeMgBr followed by HCl to convert 3 to senepodine G (2) (6 steps, 40% overall yield) and the stereospecific NaBH4 reduction of 2 to give cermizine C (1) (7 steps, 40% overall yield).
Experimental Section
3,6,7,8,9,9a-Hexahydro-2-methyl-(4H)-quinolizin-4-one (11)
Meldrum’s acid (778 mg, 5.40 mmol) and AcOH (0.26 mL, 4.5 mmol) were added in succession to a solution of pelletierine (7)5 (636 mg, 4.50 mmol) in EtOH (5 mL) and the resulting clear solution was heated to 60 °C and stirred for 1 d. 6 The yellow solution was allowed to cool; an aliquot was concentrated under reduced pressure and the 1H NMR spectrum indicated that there was a 3:1 mixture of 11 and 7 and that no Meldrum’s acid remained. Meldrum’s acid (778 mg, 5.40 mmol) was added to the yellow solution and the resulting solution was heated to 60 °C and stirred for 1d. The EtOH was removed under reduced pressure and the resulting yellowish liquid was diluted with EtOAc (30 mL). The resulting solution was washed with a saturated Na2CO3 solution (6 mL), dried over MgSO4, and concentrated under reduced pressure to give 638 mg of a yellowish liquid. Flash chromatography on silica gel (EtOAc) yielded 503 mg (68%) of 11: 1H NMR 5.30 (br s, 1), 4.85 (ddd, 1, J = 12.5, 2.4, 2.4), 3.75 (br d, 1, J = 11.0), 2.86 (br d, 1, J = 20.8), 2.83 (br d, 1, J = 20.8), 2.46 (ddd, 1, J = 12.8, 12.5, 2.5), 1.92-1.78 (m, 3), 1.70 (s, 3), 1.58-1.35 (m, 2), 1.23 (dddd, 1, J = 12.2, 12.2, 11.0, 3.7); 13C NMR 166.0, 128.9, 120.1, 58.2, 42.1, 36.3, 34.3, 25.3, 24.7, 21.8; IR (neat) 1644; HRMS (ES) calcd for C10H16NO (MH+) 166.1232, found 166.1228.
(2S*,5R)-2-Octahydro-2-methyl-(4H)-quinolizin-4-one (12)
PtO2 (8 mg, 0.03 mmol) was added to a solution of 11 (440 mg, 2.66 mmol) in EtOH (4 mL) and the resulting suspension was shaken under 50 psi of H2 for 6 h. The catalyst was removed by filtration through Celite and the resulting solution was concentrated under reduced pressure to give 425 mg (96%) of a 16:1 mixture of 12 and 3 as a clear liquid.
The spectral data for 12 were determined from the mixture: 1H NMR 4.76 (br d, 1, J = 13.8), 3.17 (dddd, 1, J = 11.0, 11.0, 6.7, 2.5), 2.45 (ddd, 1, J = 16.4, 3.4, 3.4), 2.39 (br dd, 1, J = 13.8, 12.8), 1.98-1.66 (m, 6), 1.46-1.30 (m, 2), 1.26-1.10 (m, 2), 0.97 (d, 3, J = 6.1); 13C NMR 169.4, 56.6, 41.8, 41.1, 39.5, 34.5, 26.4, 25.3, 24.2, 21.2; IR (neat) 1643. The spectral data match those previously reported for 12.7
1,6,7,8,9,9a-Hexahydro-2-methyl-(4H)-quinolizin-4-one (13)
K2CO3 (293 mg, 2.12 mmol) was added to a solution of 11 (35 mg, 0.21 mmol) in MeOH (2 mL) and the resulting suspension was stirred at 25 °C for 22 h. The suspension was filtered through Celite and MgSO4 and the resulting solution was concentrated under reduced pressure to give 35 mg of a 3:1 mixture of 13 and 11. Flash chromatography on silica gel (EtOAc) yielded 3 mg of 13, followed by 29 mg of a 3:1 mixture of 13 and 11, and 1 mg of 11.
Data for 13: 1H NMR 5.69 (s, 1), 4.48 (br d, 1, J = 13.0), 3.42-3.35 (m, 1), 2.50 (br dd, 1, J = 13.0, 12.6), 2.37 (dd, 1, J = 17.4, 6.1), 2.15 (br dd, 1, J = 17.4, 9.8), 1.90-1.65 (m, 3), 1.86 (s, 3), 1.55-1.35 (m, 3); 13C NMR 166.3, 149.0, 120.0, 54.6, 42.7, 36.3, 33.4, 24.8, 23.9, 22.7; IR (neat) 1674, 1612. The spectral data match those previously reported.7
K2CO3 (8 mg, 0.06 mmol) was added to a solution of 13 (1 mg, 0.006 mmol) in MeOH (1 mL) and the resulting suspension was stirred at 25 °C for 22 h. The suspension was filtered through Celite and MgSO4 and the resulting solution was concentrated under reduced pressure to give 1 mg of a 3:1 mixture of 13 and 11.
7-epi-Senepodine G Chloride (14)
A 1.4 M solution of MeMgBr in 3:1 toluene/THF (0.84 ml, 1.2 mmol) was added dropwise to a solution of 12 (49 mg, 0.29 mmol) in dry THF (4 mL) and the resulting solution was stirred at 60 °C for 2.5 h. The solution was allowed to cool, MeOH (5 mL) was added and the resulting solution was concentrated under reduced pressure. 3 M HCl in MeOH (2 mL) was added and the resulting solution was concentrated under reduced pressure. Flash chromatography on silica gel (70:29:1 CH2Cl2/MeOH/3 M HCl in MeOH) yielded 59 mg (100%) of 14 as a clear wax: 1H NMR (CD3OD) 4.51 (br d, 1 J = 12.5), 3.95-3.80 (m, 1), 3.43 (br dd, 1, J = 12.5, 12.2), 2.94 (br d, 1, J = 20.8), 2.58-2.40 (m, 1), 2.47 (s, 3), 2.20-2.10 (m, 2), 2.06-1.68 (m, 5), 1.57 (dddd, 1, J = 12.2, 12.2, 12.2, 4.3), 1.36 (ddd, 1, J = 12.4, 12.4, 12.4), 1.04 (d, 3, J = 6.7); 13C NMR (CD3OD) 188.8, 64.3, 54.6, 44.1, 38.5, 34.9, 26.1, 24.6, 24.1, 23.6, 20.4; HRMS (EI) calcd for C11H20N (M+) 166.1596, found 166.1599.
5-epi-Cermizine C (15)
NaBH4 (4 mg, 0.104 mmol) was added to a solution of 14 (19 mg, 0.094 mmol) in MeOH (4 mL) and the resulting suspension was stirred at 25 °C for 10 min. AcOH (~2 drops) was then added and the resulting solution was concentrated under reduced pressure. The residue was diluted with a 3:1 H2O/saturated Na2CO3 solution (4 mL) and the resulting aqueous solution was extracted with EtOAc (3 × 10 mL). The combined extracts were dried over MgSO4 and concentrated under reduced pressure to give 13 mg (82%) of 15 as a clear oil: 1H NMR (CD3OD) 3.35-3.27 (m, 1), 2.20-2.10 (m, 1), 2.02-1.92 (m, 1), 1.84 (br dd, 1, J = 11.9, 11.9), 1.76-1.50 (m, 6), 1.40-1.25 (m, 2), 1.14-0.88 (m, 3), 1.12 (d, 3, J = 6.1), 0.90 (d, 3, J = 6.1); 13C NMR (CD3OD) 64.4, 62.1, 52.5, 43.9, 42.6, 33.8, 31.4, 26.4, 24.9, 22.1, 19.8; IR (neat) 2926, 2783; HRMS (ES) calcd for C11H22N (MH+) 168.1752, found 168.1752. TFA (excess) was added to a solution of 5 in CD3OD: 1H NMR (CD3OD) 3.79 (br d, 1, J = 12.5, H1eq), 3.21-3.10 (m, 1, H9), 3.06 (br dd, 1, J = 11.3, 11.3, H5), 2.74 (ddd, 1, J = 12.8, 12.5, 2.5, H1ax), 2.15-1.65 (m, 7, contains H7), 1.62-1.48 (m, 2), 1.40-1.24 (m, 2), 1.37 (d, 3, J = 6.7), 0.98 (d, 3, J = 6.1).
(Diethoxyphosphoryl)acetyl chloride
(Diethoxyphosphoryl)acetic acid (3.001 g, 15.3 mmol) was added dropwise to thionyl chloride (3 mL) and the resulting solution was stirred at 25 °C for 1.75 h and concentrated under reduced pressure (<20 °C) to give (diethoxyphosphoryl)acetyl chloride which was used without further purification. The spectral data matched those previously reported.11
Diethyl{2-[(2S)-2-(2-hydroxyethyl)piperidin-1-yl]-2-oxoethyl}phosphonate (17)
A solution of (diethoxyphosphoryl)acetyl chloride (3.283 g, 15.3 mmol) in dry THF (6 mL) was added dropwise to a solution of (S)-2-piperdineethanol10b (659 mg, 5.10 mmol) in dry THF at 0 °C. (The detailed procedure for the resolution of 2-piperidineethanol is provided in the Supporting Information.) EtN(i-Pr)2 (2.8 mL, 16 mmol) was added dropwise to the yellow suspension and the resulting dark orange suspension was stirred at 0 °C for 1 h. The suspension was allowed to warm to 25 °C and was stirred for an additional 1 h. H2O (50 mL) was added to the suspension and the aqueous solution was extracted with EtOAc (4 × 120 mL). The combined extracts were dried over MgSO4 and concentrated under reduced pressure to 3 g of the crude ester amide as a yellowish liquid, which was used without purification.
K2CO3 (3.524 g, 25.5 mmol) was added to a solution of crude ester amide (3 g) in dry EtOH (40 mL) and the resulting suspension was heated to 60 °C and stirred for 4.5 h. The suspension was allowed to cool and excess EtOH was removed under reduced pressure. The residue was diluted in EtOAc (100 mL) and the suspension was filtered through Celite. The resulting solution was concentrated under reduced pressure to give 3 g of a dark yellow oil. Flash chromatography on silica gel (39:1 to 23:2 CH2Cl2/MeOH) yielded 1.264 g (81% from (S)-2-piperdineethanol) of 17 as a light yellow oil: [α]23 D −29 (c 1.0, CHCl3); IR (neat) 3424, 2941, 1624, 1450, 1248; HRMS (ES) calcd for C13H27NO5P (MH+) 308.1627, found 308.1638; the 1H NMR and 13C NMR spectra indicated a complex mixture of amide rotamers that match those previously reported9 and are available in the supporting information.
Diethyl {2-Oxo-2-[(2S)-2-(2-oxoethyl)piperidin-1-yl]ethyl} Phosphonate
Dess-Martin reagent (840 mg, 1.98 mmol) was added to a solution of 17 (484 mg, 1.58 mmol) in dry CH2Cl2 (15 mL) and the resulting suspension was stirred for 15 h at 25 °C. The suspension was filtered through Celite and K2CO3 and the resulting solution was concentrated under reduced pressure to 1.314 g of a white solid. Flash chromatography on silica gel (49:1 EtOAc/Et3N) yielded 383 mg (79%) of the aldehyde as a clear oil: [α]23 D −22 (c 1.0, CHCl3); the spectral data match those previously reported.9
(2S)-1,6,7,8,9,9a-Hexahydro-(4H)-quinolizin-4-one (6)
NaH (60% dispersion in oil, 43 mg, 1.1 mmol) was added to a solution of the above aldehyde (312 mg, 1.02 mmol) in dry THF (20 mL) at 0 °C and the resulting suspension was allowed to warm to 25 °C over 30 min. The orange solution was diluted with H2O (20 mL) and CH2Cl2 and the organic and aqueous layers were separated. The aqueous layer was extracted with EtOAc (3 × 80 mL) and the combined organic extracts were dried over MgSO4. The resulting solution was concentrated under reduced pressure to give 180 mg of a clear oil. Flash chromatography on silica gel (49:1 CH2Cl2/MeOH) afforded 124 mg (80%) of 6 as a clear oil: [α]23 D +47 (c 1.0, CHCl3); the spectral data match those previously reported.9
(2S,5S)-2-Octahydro-2-methyl-(4H)-quinolizin-4-one (3)
1.6 M MeLi in Et2O (4.0 mL, 6.4 mmol) was added dropwise to a suspension of CuI (625 mg, 3.28 mmol) in dry THF (20 mL) at 0 °C and the resulting solution was stirred at 0 °C for 30 min. The solution was cooled to −78 °C and BF3·OEt2 (0.41 mL, 3.2 mmol) was added dropwise. The resulting solution was stirred at −78 °C for 5 min. A solution of 6 (242 mg, 1.60 mmol) in dry THF (10 mL) was added dropwise and the resulting solution was allowed to warm to 25 °C over 20 min. Saturated NH4Cl solution (40 mL) was added and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 100 mL) and the combined extracts were dried over MgSO4. The resulting solution was concentrated under reduce pressure to give 456 mg of a yellow amorphous solid. Flash chromatography on silica gel (49:1 to 23:1 CH2Cl2/MeOH) gave 215 mg (80%) of 3 as an oil: [α]23 D −21 (c 1.0, CHCl3); 1H NMR 4.76 (dddd, 1, J = 13.1, 4.0, 2.0, 2.0), 3.37-3.29 (m, 1), 2.46 (br d, 1 J = 16.5), 2.41 (ddd, 1, J = 13.1, 12.8, 3.0), 2.11-2.00 (m, 1), 1.97 (dd, 1, J = 16.5, 9.2), 1.92-1.84 (m, 1), 1.72-1.32 (m, 7), 0.98 (d, 3, J = 6.1); 13C NMR 168.5, 55.5, 43.0, 40.6, 36.8, 33.6, 25.4, 25.0, 24.4, 20.4; IR (neat) 1639; HRMS (EI) calcd for C10H17NO (M+) 167.1310, found 167.1313.
Alternatively, TMSCl was used to promote the conjugate addition according to the following procedure: 1.6 M MeLi in Et2O (0.35 mL, 0.56 mmol) was added dropwise to a suspension of CuI (56 mg, 0.29 mmol) in dry THF (3 mL) at 0 °C and the resulting solution was stirred at 0 °C for 30 min. The solution was cooled to −78 °C and a solution of 6 (21 mg, 0.14 mmol) and TMSCl (0.035 mL, 0.28 mmol) in dry THF (2 mL) was added dropwise. The resulting solution was allowed to warm to 25 °C over 1 h. Saturated NH4Cl solution (5 mL) was added and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 10 mL) and the combined extracts were dried over Na2SO4. The resulting solution was concentrated under reduce pressure to give 30 mg of a yellow amorphous solid. Flash chromatography on silica gel (49:1 to 24:1 CH2Cl2/MeOH) gave 18 mg (77%) of a 14:1 mixture of 3 and 12 as an oil.
Senepodine G Chloride (2)
1.4 M MeMgBr in 3:1 toluene/THF (0.36 mL, 0.50 mmol) was added dropwise to 3 (21 mg, 0.13 mmol) in dry THF (4 mL) and the resulting solution was heated to 60 °C for 2.5 h. The solution was allowed to cool, MeOH (5 mL) was added and the resulting solution was concentrated under reduced pressure. 3 M HCl in MeOH (2 mL) was added and the resulting solution was concentrated under reduced pressure. Flash chromatography on silica gel (70:29:1 CH2Cl2/MeOH/3 M HCl in MeOH) yielded 25 mg (98%) of 2 as a clear wax: [α]23 D −77 (c 1.0, MeOH), lit.1 [α]26 D −35 (c 0.3, MeOH); 1H NMR (CD3OD) 4.51 (br d, 1, J = 12.8), 4.25-3.93 (m, 1), 3.55 (br dd, 1, J = 12.8, 12.8), 3.01 (dd, 1, J = 20.7, 4.3), 2.55-2.42 (m, 1), 2.46 (s, 3), 2.10-1.72 (m, 9), 1.05 (d, 3, J = 6.7); 13C NMR (CD3OD) 186.9, 64.8, 56.1, 43.9, 35.5, 34.7, 27.6, 24.7, 23.9, 21.9, 20.5; HRMS (EI) calcd for C11H20N (M+) 166.1596, found 166.1599. The spectral data match those reported for the natural product.1
Cermizine C Hydrochloride (1)
NaBH4 (5 mg, 0.1 mmol) was added to a solution of 2 (25 mg, 0.12 mmol) in MeOH (4 mL) and the resulting suspension was stirred at 25 °C for 10 min. 3 M HCl in MeOH (1 mL) was added and the resulting solution was concentrated under reduced pressure. The residue was diluted with 3:1 CH2Cl2/MeOH (10 mL) and resulting suspension was filtered to remove inorganic solids, dried over Na2SO4 and concentrated under reduced pressure to give 25 mg (99%) of 1 as a clear wax: [α]23 D −2 (c 0.4, MeOH), lit.1 [α]25 D +4 (c 0.8, MeOH); 1H NMR (CD3OD) 3.90-3.80 (m, 1), 3.68-3.55 (m, 2), 3.08 (ddd, 1, J = 13.4, 13.4, 3.0), 2.17 (dddd, 1, J = 13.4, 13.4, 13.4, 4.3), 2.06-1.54 (m, 9), 1.38-1.20 (m, 1), 1.33 (d, 3, J = 6.1), 0.95 (d, 3, J = 6.7); 13C NMR (CD3OD) 61.3, 51.2, 49.9, 41.7, 38.3, 25.4, 24.6, 23.8, 21.6, 18.4, 17.7; HRMS (EI) calcd for C11H21N (M+) 167.1673, found 167.1674. The spectral data match those reported for the natural product.1
Supplementary Material
. General procedures, details of NOE experiments on 15, and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
SCHEME 2.

Synthesis of 5-epi-Cermizine C (15) and 7-epi-Senepodine G (14)
Acknowledgments
We thank the NIH (GM50151) for generous financial support.
References and Notes
- 1.Morita H, Hirasawa Y, Shinzato T, Kobayashi J. Tetrahedron. 2004;60:7015–7023. [Google Scholar]
- 2.For the isolation of other members of these families see: Morita H, Hirasawa Y, Yoshida N, Kobayashi J. Tetrahedron Lett. 2001;42:4199–4201.Hirasawa Y, Morita H, Kobayashi J. Tetrahedron. 2003;59:3567–3573.Hirasawa Y, Morita H, Kobayashi J. Heterocycles. 2004;64:515–521.
- 3.For a review of lycopodium alkaloids see: Ma X, Gang DR. Nat Prod Rep. 2004;21:752–772. doi: 10.1039/b409720n.
- 4.Bohlmann F, Winterfeldt E, Boroschewski G, Mayer-Mader R, Gatscheff B. Chem Ber. 1963;96:1792–1800. [Google Scholar]
- 5.Quick J, Oterson R. Synthesis. 1976:745–746. [Google Scholar]
- 6.(a) Meghani P, Joule JA. J Chem Soc, Perkins Trans 1. 1988:1–8. [Google Scholar]; (b) Janecki T, Bodalski R, Wieczorek M, Bujacz G. Tetrahedron. 1995;51:1721–1740. [Google Scholar]
- 7.(a) Ban Y, Kimura M, Oishi T. Heterocycles. 1974;2:323–328. [Google Scholar]; (b) Ban Y, Kimura M, Oishi T. Chem Pharm Bull. 1976;24:1490–1496. [Google Scholar]
- 8.In 1935, Ochiai reported the addition of MeMgI to 12 or its diastereomer, followed by high pressure hydrogenation to give 14 or a diastereomer: Ochiai E, Tsuda K, Yokoyama J. Chem Ber. 1935;68:2291–2298.For a related conversion of a lactam to a methyl quinolizidine with MeMgBr and NaBH3CN see: Comins DL, Zheng Z, Goehring RR. Org Lett. 2002;4:1611–1613. doi: 10.1021/ol025820q.
- 9.(a) Grzyb JA, Batey RA. Tetrahedron Lett. 2003;44:7485–7488. [Google Scholar]; (b) Grzyb JA, Shen M, Yoshina-Ishii C, Chi W, Brown RS, Batey RA. Tetrahedron. 2005;61:7153–7175. [Google Scholar]
- 10.(a) Toy MS, Price CC. J Am Chem Soc. 1960;82:2613–2616. [Google Scholar]; (b) Beyerman HC, Van Den Bosch S, Breuker JH, Maat L. Recl Trav Chim Pays-Bas. 1971;90:755–764. [Google Scholar]; (c) Craig JC, Lee S-YC, Pereira WE, Jr, Beyerman HC, Maat L. Tetrahedron. 1978;34:501–504. [Google Scholar]
- 11.Teichert A, Jantos K, Harms K, Studer A. Org Lett. 2004;6:3477–3480. doi: 10.1021/ol048759t. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yamamoto Y, Yamamoto S, Yatagai H, Ishihara Y, Maruyama K. J Org Chem. 1982;47:119–126. [Google Scholar]; (b) Smith AB, III, Jerris PJ. J Am Chem Soc. 1981;103:194–195. [Google Scholar]
- 13.(a) Matsuzawa S, Horiguchi Y, Nakamura E, Kuwajima I. Tetrahedron. 1989;45:349–362. [Google Scholar]; (b) Nakamura E, Yamanaka M, Mori S. J Am Chem Soc. 2000;122:1826–1827. [Google Scholar]; (c) Alexakis A, Berlan J, Besace Y. Tetrahedron Lett. 1986;27:1047–1050. [Google Scholar]
- 14.Nizamkhodzhaeva AN, Ishbaev AI, Aslanov Kh A, Mukhamedzhanov SZ. Chem Abstr. 1978;88:121499v. [Google Scholar]
- 15.Moynehan TM, Schofield K, Jones RAY, Katritzky AR. J Chem Soc. 1962:2637–2658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. General procedures, details of NOE experiments on 15, and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org