

Abstract
Aims/hypothesis
Mitochondrial dysfunction has been postulated to underlie muscular fat accumulation, leading to muscular insulin sensitivity and ultimately type 2 diabetes mellitus. Here we re-interpret previously published data on [13C]acetate recovery in breath gas obtained during exercise in type 2 diabetic patients and control individuals.
Methods
When infusing [13C]palmitate to estimate fat oxidation, part of the label is lost in exchange reactions of the tricarboxylic acid (TCA) cycle. To correct for this loss of label, an acetate recovery factor (ARF) has previously been used, assuming that 100% of the exogenously provided acetate will enter the TCA cycle. The recovery of acetate in breath gas depends on the TCA cycle activity, hence providing an indirect measure of the latter and a marker of mitochondrial function.
Results
Re-evaluation of the available literature reveals that the ARF during exercise is highest in lean, healthy individuals, followed by obese individuals and type 2 diabetic patients.
Conclusions/interpretation
Revisiting previously published findings on the ARF during exercise in type 2 diabetic patients reveals a reduction in muscular TCA cycle flux, reflecting mitochondrial dysfunction, in these patients. How mitochondrial dysfunction is related to type 2 diabetes mellitus—cause or consequence—requires further study.
Keywords: Human, Mitochondria, Mitochondrial function, Muscle, Stable isotopes, Type 2 diabetes mellitus
The prevalence of type 2 diabetes mellitus is increasing rapidly and is affecting the health of millions of humans now, and will do so in the near future. Among the factors that are responsible for the increasing prevalence of this disease are obesity, consumption of energy-dense diets and low levels of physical activity. Together, these factors contribute to fat accumulation in non-adipose tissues, like heart, liver and skeletal muscle. Indeed, the accumulation of fat in skeletal muscle is an early hallmark of the development of type 2 diabetes mellitus, and high muscular fat levels can already be detected in the prediabetic state [1]. In the last 5 years, a reduced muscular oxidative capacity, referred to as mitochondrial dysfunction, has been postulated to contribute to fat accretion in skeletal muscle. Thus, reduced in vivo mitochondrial function under resting conditions as well as during post-exercise recovery has been observed in type 2 diabetic patients [2] and in first-degree relatives of type 2 diabetic patients [3], and a reduced mitochondrial tricarboxylic acid (TCA) cycle flux was shown to underlie the reduced mitochondrial oxidative capacity in insulin-resistant offspring of type 2 diabetic patients [4]. But other studies have reported no defects in mitochondrial function in diabetic patients [5, 6]. The reason for this discrepancy in findings is so far unclear, but could include differences in individual characteristics, the severity and duration of the patients’ type 2 diabetes, the type and duration of the glucose-lowering medication used, and their habitual physical fitness levels.
Therefore, the current debate on mitochondrial dysfunction as a contributor to the pathogenesis of type 2 diabetes is ongoing and has called for more studies towards mitochondrial function in patients with type 2 diabetes. These studies may, however, be biased by prior knowledge of reported defects in mitochondrial function. We here re-evaluate existing data that was gathered for other goals and from another perspective and in a period when mitochondrial dysfunction had not yet been proposed to be key in the development of type 2 diabetes. This unbiased approach has led us to conclude that a reduced TCA cycle flux is part of the defect in mitochondrial dysfunction observed in type 2 diabetes.
In 1995, the acetate recovery factor (ARF) was introduced in the field of stable isotope tracer methodology, to correct tracer estimates of fat oxidation for the loss of label in exchange reactions of the TCA cycle [7]. Thus, when [13C]palmitate is used to determine plasma-derived palmitate oxidation, part of the infused label is lost in exchange reactions of the TCA cycle (Fig. 1). To correct for this loss of label, the appearance of the 13C label of [13C]acetate into 13CO2 can be used. Acetate is immediately converted into acetyl coenzyme A (acetyl-CoA), and it is assumed that almost 100% of the labelled [13C]acetate will enter the TCA cycle. Under lipogenic conditions, though, it cannot be excluded that some of the infused acetate is used for de novo lipogenesis. However, under normal resting conditions only a fraction of the infused [13C]acetate is recovered as 13CO2 in breath gas, indicating that indeed most of the 13C label is lost in the TCA cycle. Interestingly, the loss of label in the TCA cycle depends on the flux of the TCA cycle. Thus, the recovery of acetate as 13CO2 in breath gas is only about 20% during rest, but increases up to about 100% during high-intensity exercise, when skeletal muscle mitochondrial TCA activity is accelerated and de novo lipogenesis is negligible. Indeed, we previously showed that the recovery of acetate during exercise is highly dependent on metabolic rate [8]. Especially during exercise and when measured under standardised conditions, the ARF can provide a good, albeit indirect, indication of TCA cycle flux in skeletal muscle mitochondria.
Fig. 1.

Metabolic routes of [13C]acetate. The 13C label can be lost in exchange reactions of the TCA cycle, which will reduce the recovery of the label in 13CO2 in breath gas. The loss of 13C label in exchange reactions depends on the rate of the TCA cycle flux
With the above in mind, it is very interesting to note that the ARF has been previously determined under standardised conditions in (pre)-diabetic patients compared with control individuals, albeit not with the original aim of using this factor as an indirect measure of mitochondrial TCA cycle flux. Thus, Borghouts et al. [9] reported that male, overweight, type 2 diabetic patients (see Table 1 for individual characteristics) had a 23% lower recovery of acetate during exercise (performed at about 40–45% of maximal aerobic capacity [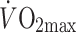 ]) when compared with body weight-matched healthy controls (ARF: 54.5 ± 0.06% vs 70.7 ± 0.07%, p < 0.05). Because
]) when compared with body weight-matched healthy controls (ARF: 54.5 ± 0.06% vs 70.7 ± 0.07%, p < 0.05). Because 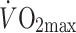 was different between controls and diabetic patients in this study, the latter group exercised at the same relative but a somewhat lower absolute workload, which may explain part of the difference in ARF. However, Mensink et al. [10] measured acetate recovery during exercise (at 55–58%
was different between controls and diabetic patients in this study, the latter group exercised at the same relative but a somewhat lower absolute workload, which may explain part of the difference in ARF. However, Mensink et al. [10] measured acetate recovery during exercise (at 55–58% 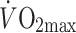 ) in male obese type 2 diabetic patients, individuals with an impaired glucose tolerance (IGT) and healthy controls, all matched for percentage body fat and
) in male obese type 2 diabetic patients, individuals with an impaired glucose tolerance (IGT) and healthy controls, all matched for percentage body fat and 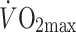 (Table 1). Again, acetate recovery tended to be lower in type 2 diabetic patients and IGT individuals compared with healthy controls (about 63, 63 and 71%, in type 2 diabetes mellitus, IGT and controls, respectively, n = 7, p = 0.07). When jointly examining ARFs obtained during exercise under standardised conditions in our laboratory, at on average 50% of
(Table 1). Again, acetate recovery tended to be lower in type 2 diabetic patients and IGT individuals compared with healthy controls (about 63, 63 and 71%, in type 2 diabetes mellitus, IGT and controls, respectively, n = 7, p = 0.07). When jointly examining ARFs obtained during exercise under standardised conditions in our laboratory, at on average 50% of 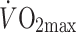 , we previously demonstrated that the ARF was highest in lean, healthy individuals (78.5 ± 2.1%, n = 21), followed by obese individuals (69.9 ± 1.7%, n = 31) and type 2 diabetic patients (59.2 ± 2.1%, n = 17) [8]. Remarkably, even after correction for factors related to mitochondrial function, which actually may differ between diabetic patients and control individuals, like RQ, obesity (as defined by body fat%) and metabolic rate (workload), the ARF tended to be lower in type 2 diabetic patients compared with obese and lean controls [8]. This further strengthens the unbiased observation of a reduced TCA cycle flux in type 2 diabetic patients.
, we previously demonstrated that the ARF was highest in lean, healthy individuals (78.5 ± 2.1%, n = 21), followed by obese individuals (69.9 ± 1.7%, n = 31) and type 2 diabetic patients (59.2 ± 2.1%, n = 17) [8]. Remarkably, even after correction for factors related to mitochondrial function, which actually may differ between diabetic patients and control individuals, like RQ, obesity (as defined by body fat%) and metabolic rate (workload), the ARF tended to be lower in type 2 diabetic patients compared with obese and lean controls [8]. This further strengthens the unbiased observation of a reduced TCA cycle flux in type 2 diabetic patients.
Table 1.
Characteristics of the individuals
| Controls | Type 2 diabetes | IGT | |
|---|---|---|---|
| Borghouts et al. [9] | |||
| n | 8 | 8 | – |
| Age (years) | 47 ± 5 | 58 ± 5 | – |
| Body weight (kg) | 86.3 ± 9.5 | 86.4 ± 14.1 | – |
| Body fat (%) | 29.2 ± 5.3 | 26.5 ± 8.4 | – |
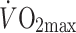 (ml kg−1min−1) (ml kg−1min−1) |
36.2 ± 2.0 | 25.8 ± 3.3 | – |
Exercise intensity (% of 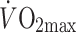 ) ) |
44 ± 3 | 41 ± 6 | – |
| Mensink et al. [10] | |||
| n | 7 | 7 | 7 |
| Age (years) | 45.1 ± 4.5 | 51.3 ± 9.0 | 58.3 ± 6.3 |
| Body weight (kg) | 100.8 ± 9.8 | 93.0 ± 8.7 | 110.8 ± 18.8 |
| Body fat (%) | 35.0 ± 3.2 | 36.6 ± 3.4 | 33.0 ± 3.2 |
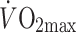 (ml [kg FFM]−1 min−1) (ml [kg FFM]−1 min−1) |
41.5 ± 3.2 | 36.2 ± 7.7 | 39.6 ± 2.4 |
Exercise intensity (% of 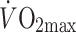 ) ) |
57.8 ± 9.5 | 55.6 ± 5.6 | 54.9 ± 12.2 |
Values are means ± SD
FFM, fat-free mass
Although our re-evaluation of previously published data points towards a reduced TCA cycle flux in type 2 diabetic patients, it cannot answer the question of whether mitochondrial dysfunction is the cause or consequence of type 2 diabetes mellitus. The finding that the ARF was reduced both in type 2 diabetic patients and in prediabetic individuals with IGT—comparable with the findings of reduced in vivo mitochondrial function in first-degree relatives [4]—could be interpreted as evidence for a causal relationship between mitochondrial dysfunction and the development of diabetes. However, in these studies, (pre)diabetic individuals are characterised by insulin resistance and associated high insulin levels. In that respect, a recent study showed that in type 2 diabetic patients, high insulin levels reduced the expression of peroxisome proliferator-activated receptor-gamma coactivator-1α, citrate synthase and cytochrome oxidase subunit 1 and blunted the insulin-stimulated increase in mitochondrial ATP production [11]. In addition, we observed in patients with type 2 diabetes that mitochondrial function, measured as post-exercise phosphocreatine resynthesis rate, correlated inversely with fasting blood glucose and HbA1c levels [2]. Therefore, mitochondrial dysfunction could also very well reflect a consequence of the insulin-resistant state. Alternatively, a reduced TCA cycle flux in type 2 diabetic patients may be the consequence of a more sedentary, inactive lifestyle. So far, most studies that report mitochondrial dysfunction in type 2 diabetes have not carefully matched for habitual levels of physical (in)activity. It is well known that physical endurance training is one of the most powerful strategies to prevent and treat type 2 diabetes mellitus and it is not unlikely that training can do so by improving mitochondrial function.
In conclusion, revisiting previously published findings on the ARF during exercise in type 2 diabetic patients reveals a reduction in muscular TCA cycle flux in these patients. Why ARF is reduced in type 2 diabetic patients was previously unexplained, but now provides an unbiased additional indication that indeed mitochondrial function is reduced in type 2 diabetic patients. How mitochondrial dysfunction is related to type 2 diabetes mellitus—cause or consequence—requires further study.
Acknowledgements
The research of P. Schrauwen was made possible by a fellowship of the Royal Netherlands Academy of Arts and Sciences. The research of M. K. C. Hesselink was made possible by a Vidi grant for innovative research (no. 917.66.359).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- ARF
acetate recovery factor
- acetyl-CoA
acetyl coenzyme A
- IGT
impaired glucose tolerance
- TCA
tricarboxylic acid
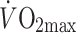
maximal aerobic capacity
References
- 1.Perseghin G, Scifo P, De Cobelli F, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48:1600–1606. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- 2.Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, et al. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007;50:113–120. doi: 10.1007/s00125-006-0475-1. [DOI] [PubMed] [Google Scholar]
- 3.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Befroy DE, Petersen KF, Dufour S, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair KS, Bigelow ML, Asmann YW, et al. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes. 2008;57:1166–1175. doi: 10.2337/db07-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR. A new correction factor for use in tracer estimations of plasma fatty acid oxidation. Am J Physiol. 1995;269:E649–E656. doi: 10.1152/ajpendo.1995.269.4.E649. [DOI] [PubMed] [Google Scholar]
- 8.Schrauwen P, Blaak EE, Van Aggel-Leijssen DP, Borghouts LB, Wagenmakers AJ. Determinants of the acetate recovery factor: implications for estimation of [13C]substrate oxidation. Clin Sci (Lond) 2000;98:587–592. [PubMed] [Google Scholar]
- 9.Borghouts LB, Wagenmakers AJ, Goyens PL, Keizer HA. Substrate utilization in non-obese Type II diabetic patients at rest and during exercise. Clin Sci (Lond) 2002;103:559–566. doi: 10.1042/cs1030559. [DOI] [PubMed] [Google Scholar]
- 10.Mensink M, Blaak EE, van Baak MA, Wagenmakers AJ, Saris WH. Plasma free fatty acid uptake and oxidation are already diminished in subjects at high risk for developing type 2 diabetes. Diabetes. 2001;50:2548–2554. doi: 10.2337/diabetes.50.11.2548. [DOI] [PubMed] [Google Scholar]
- 11.Asmann YW, Stump CS, Short KR, et al. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–3319. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]