
Figure 4. Comparison of GOLD predicted binding geometry of natural pentasaccharide H5 having ‘average backbone’ with that of H5CRYS determined in the crystal structure.
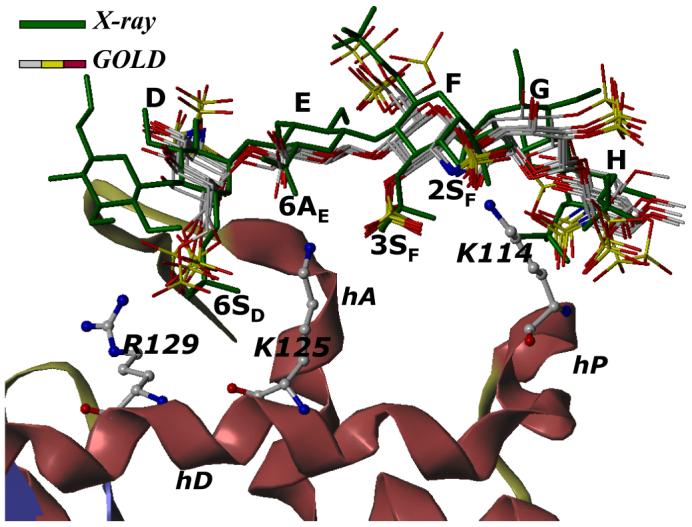
H5 with average inter-glycosidic bond parameters (‘the average backbone’), was docked onto antithrombin. Structurally H5 differs from H5CRYS (see Fig. 1). An overlay of 6 solutions from three independent docking runs shows high consistency in the predicted binding geometry, which matches the crystal structure geometry with an RMSD less than 2.5 Å. Structure in green is the crystal structure geometry, while those in atom-type color (red, yellow, grey and blue) are 6 docking solutions. Note the identical orientation of key groups, 2- and 3-OSO3- of residue F (2SF and 3SF), 6-COO- of residue E (6AE) and 6-OSO3- of residue D (6SD). Helices A, D and P of antithrombin (in ribbon diagram) are indicated as hA, hD and hP, while D, E, F, G, and H labels correspond to residues of the pentasaccharide. K114, K125 and R129 are shown in ball and stick representation. See text for details.