

Abstract
The adipose tissue-derived hormone, leptin, acts via its receptor (LepRb) in the brain to regulate energy balance and neuroendocrine function. Parsing the biology of leptin requires understanding LepRb signaling and the roles for specific signaling pathways in neural and physiological leptin action. Since the leptin acts via a broadly distributed network of LepRb-expressing neurons, understanding the function of each of these LepRb neural populations will also be crucial. Here, we review the status of knowledge regarding the molecular mediators of leptin action and the neural substrate via which leptin acts to regulate physiologic processes.
Leptin
Adipose tissue produces the hormone leptin in approximate proportion to fat stores. Circulating leptin communicates the level of energy reserves in the periphery to the central nervous system (CNS) in order to suppress food intake and permit energy expenditure (1–4). Adequate leptin levels permit energy expenditure on the processes of reproduction and growth, and similarly regulate other elements of the endocrine and immune systems (4–6). Conversely, lack of leptin signaling due to mutation of leptin (e.g. ob/ob mice) or the leptin receptor (LepR) (e.g. db/db mice) in rodents and humans results in increased food intake in combination with reduced energy expenditure (and thus obesity), plus neuroendocrine dysfunction (including hypothyroidism, decreased growth, infertility and decreased immune function) (1;7–9). Many of the effects of leptin are attributable to effects in the CNS, particularly in the hypothalamus, a site of high LepRb mRNA expression (2;3).
The Leptin Receptor
Leptin possesses a four-helix-bundle structure characteristic of the class-I family of cytokines (10); LepR, similarly, represents a typical class-I cytokine receptor (11). Alernative splicing generates several isoforms of LepR with identical ligand binding domains, but which possess differing perimembrane and intracellular domains (12). Membrane-bound LepRs consist of long (LepRb) and short (LepRa, among others) isoforms LepRb features an approximately 300 amino acid intracellular tail that contains several docking sites for proteins critical for signal transduction. In contrast, short-form receptors contain 30–40 amino acid intracellular domains lacking these sites and which therefore play no major role in signal transduction. Consistent with the unique presence of signaling moieties on LepRb, db/db mice that lack only the LepRb isoform of the receptor phenocopy leptin-deficient ob/ob animals (12). Potentially important roles for membrane bound short-form receptors include the endocytosis and transport of leptin across the blood-brain barrier (13). Alternative splicing and proteolytic cleavage events also produce circulating extracellular domain of LepR, which may affect the stability and/or availability of circulating leptin (14;15).
LepRb and Jak2
Like other class-I cytokine receptors, LepRb has no intrinsic enzymatic activity; propagation of downstream leptin signals requires the LepRb-associated tyrosine kinase, Jak2 (16). Membrane-proximal residues on the intracellular tail of LepR including the proline-rich “Box 1” motif and the downstream “Box 2” mediate Jak2 interactions; while LepRa and other short forms contain Box 1, they do not possess Box2 and poorly interact with Jak2 (16;17) (Figure 1).
Figure 1. The role of discreet LepRb functional sites in leptin signaling.
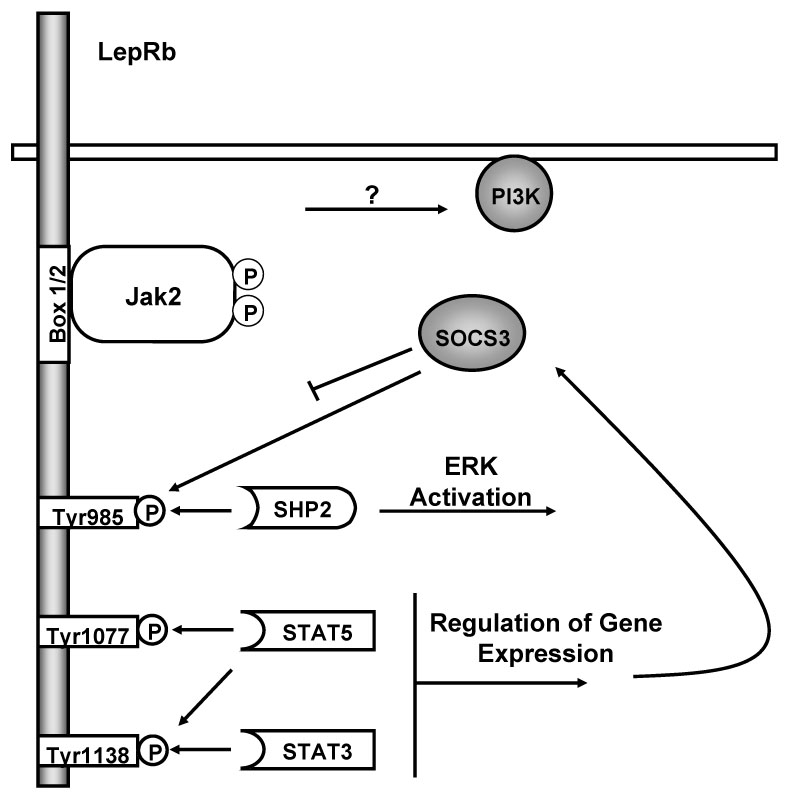
Leptin binding to LepRb activates the associated Jak2 tyrosine kinase bound at the Box 1/2 motifs. Activated Jak2 undergoes robust autophosphorylation and phosphorylates Tyr985, Tyr1077 and Tyr1138 on the LepRb intracellular tail. These phosphorylated residues act as docking sites for SH2-domain containing proteins. Phosphorylated Tyr985 mediates docking with SHP2 and subsequent activation of ERK through the MAPK signaling cascade. Phosphorylated Tyr1077 mediates STAT5 activation. Phosphorylated Tyr1138 mediates both STAT3 and STAT5 activation. STAT3 activation ultimately leads to increased expression of SOCS3, which acts as a feedback inhibitor and negatively regulates LepRb signaling in part by binding phosphorylated Tyr985. Leptin also activates PI3-K, although the intermediate steps for this process remain obscure.
Leptin binding stimulates the autophosphorylation of Jak2 on two key tyrosine residues in the activation loop of the kinase domain that promote its enzymatic activation (18;19). While the precise mechanism by which leptin triggers Jak2 activation is not completely understood, a growing body of work supports a model in which leptin binding promotes LepR aggregation into oligomers in such a way as to bring their constitutively-bound Jak2 molecules close to one another to trans-autophosphorylate (20–22).
LepRb signals require Jak kinase activity and the phosphorylation of tyrosine residues on LepRb and/or other signaling proteins. Not surprisingly, Jak2 also serves as a key point of regulation for many factors that influence flux through the LepRb signaling, including an increasingly complex set of phosphorylation events on Jak2 itself (23). In addition to the activating phosphorylation of Tyr1007 and Tyr1008 in the activation loop, Tyr1007 also plays a role in the recruitment of inhibitory proteins such as SOCS-1 and SOCS-3 (24;25). Phosphorylation at Tyr813 recruits SH2B1, an SH2-domain containing protein that augments the kinase function of Jak2, and which may mediate certain downstream signals (26). Phosphorylation of Tyr119 disrupts Jak2/cytokine receptor interaction, and phosphorylation of Ser523 and Tyr570 inhibits Jak2 kinase activity (27–30). Other regulatory Jak2 phosphorylation events certainly exist, but remain to be elucidated.
LepRb tyrosine phosphorylation mediates important downstream signals
Once activated, Jak2 phosphorylates all three conserved tyrosine residues on the intracellular tail of LepRb: Tyr985, Tyr1077, and Tyr1138, promoting their recruitment of downstream signaling proteins (31). The family of signal transducers and activators of transcription (STATs) represent the canonical Jak2/cytokine receptor-dependent signaling proteins (32). These latent transcription factors are recruited to activated cytokine receptor/Jak kinase complexes, whereupon tyrosine phosphorylation stimulates their nuclear translocation and transcriptional activation. LepRb recruits multiple STAT isoforms, and leptin stimulates the phosphorylation and activation of STAT3 and STAT5 in vivo (31;33;34).
Phosphorylation of Tyr985 recruits at least two different proteins: SHP-2 (SH2-domain containing phosphatase-2), and SOCS-3 (suppressor of cytokine signaling-3): The consensus phosphotyrosine recognition sequences of these two proteins are very similar (35). SHP-2 binds to phosphorylated Tyr985 (36;37) to mediate the activation of ERK in cultured cells (38). SOCS-3, a member of a family of SOCS-box containing proteins that attenuate cytokine signaling, mediates feedback inhibition of LepRb signaling by binding to Tyr985 (37).
In order to understand the contribution of LepRb Tyr985 to leptin action and inhibition in vivo, we generated mice in which LepRbL985 (containing a substitution of Tyr985 that abrogates phosphorylation of the site and prevents SHP2/SOCS3 binding) homologously replaced normal LepRb (38–40) (41). Mutation of Tyr985 in these mice results in reduced feeding and adiposity, decreased orexigenic ARC neuropeptide expression, and increased baseline STAT3 activation in mice-all in the face of low leptin levels. Coupled with the increased sensitivity of LepRbL985 animals to exogenous leptin, these observations suggest that mutation of Tyr985 blocks the activation of an inhibitory Tyr985-dependent LepRb signal, ultimately leading to increased leptin sensitivity in vivo. These results suggest an important role for Tyr985 in the attenuation of leptin action in vivo, consistent with results from cultured cells suggesting an important role for Tyr985 in the inhibition of LepRb signaling (33;37;40).
Since Tyr985 of LepRb recruits both SHP-2 and SOCS3 (36;37;40), the failure of LepRbL985 to recruit either of these proteins could theoretically underlie the lean, leptin-sensitive phenotype of LepRbL985 mice. Most data from cultured cells and animals support a primary role for SOCS3 in the inhibition of LepRb signaling, however (38;40;42–46).
The phosphorylation of LepRb Tyr1077 has been demonstrated only recently; indeed, it was not clear initially that this site was phosphorylated, since mutating Tyr985 and Tyr1138 eliminated all immunoreactivity of LepRb with standard anti-phosphotyrosine antibodies (36;39). The demonstration of a role for Tyr1077 in the activation of STAT5 (31;47) prompted us to re-examine this residue using a site-and phosphorylation state-specific antibody, however, revealing its ligand-dependent phosphorylation (31). The role for this site in the regulation of physiology is not yet clear.
While Tyr1077 mediates the majority of LepRb-dependent STAT5 phosphorylation and transcriptional activation, Tyr1138 also contributes to the phosphorylation of STAT5-although the importance of Tyr1138 in the transcriptional activation of STAT5 is unclear (31;47). LepRb Tyr1138 lies in a consensus YXXQ binding site for STAT3 (48), and is the sole residue responsible for the activation of STAT3 during leptin action (31). LepRb activation initiates feedback inhibition of leptin signaling by promoting the transcription of SOCS-3 in a Tyr1138/STAT3-dependent manner. Thus, in addition to blocking positive STAT3-dependent leptin actions, mutation of Tyr1138 eliminates LepRb-mediated SOCS-3 accumulation and blocks the attenuation of Jak2 and STAT5 signaling during prolonged receptor stimulation (42).
The physiological importance of Tyr1138 has been demonstrated using a knock-in mouse model in which LepRbS1138 (mutant for Tyr1138) replaces endogenous LepRb. These mice are hyperphagic and obese but lack some of the phenotypes that characterize the ob/ob and db/db models including severe diabetes and infertility (49;50). Furthermore, some leptin actions, including some aspects of immune function, are enhanced in Tyr1138-mutant mice compared to wild-type mice (42;51). Thus, while LepRb Tyr1138→.STAT3 signaling is crucial for the regulation of energy homeostasis, other LepRb signals also contribute to leptin action, and Tyr1138 (along with Tyr985) may attenuate LepRb signaling in vivo.
Other Downstream Events
While STAT3, STAT5, and ERK activation can be conveniently assigned to specific phosphorylation sites on LepRb, some downstream effects remain poorly understood. These signals include activation of phosphoinositide-3 kinase (PI3-K) and the mammalian target of Rapamycin (mTOR), and the inhibition of the AMP-activated protein kinase (AMPK) in the arcuate nucleus of the hypothalamus (ARC) (52–54). The integrity and activity of these pathways play a crucial role in leptin action in vivo: For instance, leptin increases PI 3-K activity in the ARC, and PI3-K inhibitors administered directly to the brain abrogate leptin-mediated changes in food intake and glucose homeostasis (52;55;56). The signaling events linking LepRb activation to PI3-K activation remain unclear, though some studies suggest roles for the insulin receptor substrate-(IRS)-proteins in the regulation of PI3-K activity during leptin signaling (52). Furthermore, at least in cultured cells, the Jak2-interacting protein SH2B1 interacts with Jak2 and IRS-2 and promotes IRS-1/2 mediated activation of PI3-K in response to leptin (57). While this may not be the only protein connecting LepRb/Jak2 activation to PI3-K activation, SH2B1 loss-of-function leads to significant metabolic defects in mice (58;59).
Leptin regulates neurophysiology via LepRb in the brain
LepRb-expressing neurons in the brain mediate most leptin action (60–62) (Figure 2). The largest populations of LepRb neurons reside in hypothalamic nuclei, including the arcuate (ARC), dorsomedial (DMH), ventromedial (VMH), lateral hypothalamic area (LHA), and ventral premammillary (PMv) nuclei (63–66). Additional important populations of LepRb neurons reside outside the hypothalamus, however, including in the ventral tegmental area (VTA), raphe nuclei, brainstem, periaqueductal gray matter and elsewhere.
Figure 2. Neuroanatomically discrete populations of LepRb neurons mediate distinct components of leptin action.
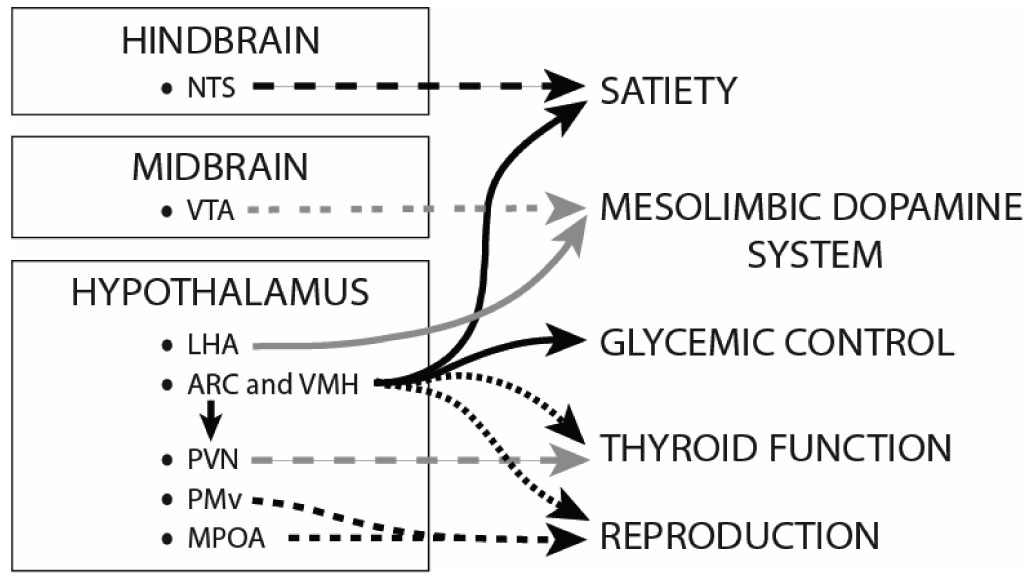
Clusters of LepRb-expressing neurons in the ARC and VMH contribute to the control of various functions, including satiety and glycemic control, and likely also affect thyroid and reproductive functions, perhaps via indirect connections with other areas. The hindbrain, including the NTS, encodes much of satiety, and NTS LepRb neurons may contribute to this effect of leptin. Leptin regulates thyroid function indirectly, at least in part via connections from ARC LepRb neurons to the PVN. Populations of LepRbexpressing neurons in the MPOA and the PMv could play a role in the modulation of the neuroendocrine reproductive axis. Leptin also regulates the mesolimbic dopamine system via LepRb-expressing neurons in the VTA and perhaps the LHA; this likely modulates the incentive value of food. Only by defining the neuronal and physiologic functions of all LepRb neurons in the brain will we understand the sum of leptin action.
LepRb action in the ARC
In the ARC, two well-characterized populations of neurons express LepRb (although others likely exist, as well): one population synthesizes the orexigenic neuropeptides agouti-related peptide (AgRP) and neuropeptide Y (NPY); the other neural population synthesizes the anorexigenic pro-hormone proopiomelanocortin (POMC) (63;67). Leptin activates/depolarizes LepRb/POMC neurons and increases POMC synthesis (67;68) to decrease appetite and increase energy expenditure by activating CNS melanocortin receptors (69–74). By contrast, leptin inhibits NPY/AgRP neurons and suppresses expression of these orexigenic neuropeptides (67;68). Collectively, leptin/LepRb modulates these ARC neurons to respond to alterations in energy homeostasis. When energy stores are high and leptin is abundant, LepRb signaling stimulates the production of anorectic POMC and suppresses levels of orexigenic AgRP and NPY. Conversely, decreased or deficient leptin activity (e.g. during starvation and in ob/ob and db/db mice) stimulates appetite by suppressing synthesis of anorectic neuropeptides and increasing expression of orexigenic peptides.
Neuropeptide Regulation
The LepRb-proximal signaling events enumerated above initiate a long chain of events that result in physiological changes; the regulation of neuropeptide gene expression represents one important early link in this chain. In the ARC, STAT3 and Foxo1 play crucial roles in neuropeptide gene regulation. Foxo1, a forkhead-family transcription factor, regulates gene expression in a PI3-K dependent manner (75): PI3-K promotes Akt activity, and the Akt-dependent phosphorylation of Foxo1 leads to its nuclear exclusion and proteosomal degradation. Studies both in cell culture and in vivo demonstrate the key role for STAT3 in the regulation of POMC expression by leptin (49;76–78). Data from LepRbS1138 mice suggest that leptin regulates AgRP through both STAT3 dependent and independent mechanisms, while STAT3 appears to play no role in the regulation of NPY expression (49). Foxo1 has been shown to bind both the NPY and AgRP promotors and stimulate them both, however, while PI3-K inhibits the expression of both (78;79). These studies suggest a model in which STAT3 primarily regulates POMC, STAT3 and Foxo1 each modulate AgRP expression, and Foxo1 modulates NPY expression independently of STAT3 in the ARC.
LepRb action beyond the ARC
While much attention has focused on understanding leptin regulation of ARC LepRb-expressing neurons, the ARC does not explain the totality of leptin’s neurophysiological effects. For example, although ablation of AgRP neurons results in hypophagia and ablation of POMC or central melanocortin receptors results in severe obesity (70;80), deletion of LepRb from POMC and/or AgRP neurons or the restoration of LepRb in the ARC of db/db animals results in only modest alteration in body weight (81–83). Furthermore, although interference with LepRb→STAT3 signaling in LepRbS1138 mice results in dramatic hyperphagia and obesity, deletion of STAT3 in POMC and NPY/AgRP neurons only modestly impacts body energy homeostasis (49;84–86). So, while the melanocortins and ARC POMC and NPY/AgRP neurons promulgate powerful appetitive signals, they probably do not mediate the majority of the leptin-mediated anorectic signal. Furthermore, LepRb-expressing neurons lie in numerous areas outside of the ARC, and ARC LepRb neurons comprise only 15–20% of the total number of LepRb-expressing neurons within the CNS (66). Presumably, the distribution LepRb-expressing neurons throughout the brain reflects the existence of multiple distinct neuronal populations, each of which mediates a specific aspect or aspects of leptin action. This arrangement represents an attractive means to explain how leptin may orchestrate diverse neural processes ranging from neuroendocrine and sympathetic nervous system function, to satiety, and to the perception of food reward.
Leptin Regulation of Satiety: the ARC, VMH and Hindbrain
Satiety is the perception of fullness that terminates feeding. The ARC and VMH are defined as “satiety centers” because lesion of either blunts satiety and promotes hyperphagia and obesity (2;3). The role of the ARC in leptin-mediated satiety, (e.g. via LepRb/POMC and LepRb/NPY) is well understood, as discussed above. Additionally, VMH LepRb neurons contribute to satiety via excitatory projections onto ARC POMC neurons (87). The density of these projections are dynamically regulated by leptin availability (i.e. fasting or fed states) demonstrating the exquisite sensitivity of the VMH to physiological changes (88). Leptin activates a subpopulation of VMH LepRb neurons co-expressing the transcription factor SF-1 that contribute to leptin-mediated satiety (89), but the roles of other VMH LepRb neurons are less clear.
The brainstem is also important in controlling satiety, particularly the nucleus of the solitary tract (NTS) and nearby interconnected regions (2;3;90). The brainstem receives numerous inputs from the gut and relays this information to hypothalamic satiety and feeding centers. Leptin and the anorexigenic gut peptides GLP-1 and cholecystokinin (CCK) act synergistically to regulate the neurons of the NTS, and thus contribute to satiety (91–93). While the NTS contains LepRb neurons, whether these hindbrain LepRb neurons (as opposed to hypothalamic LepRb neurons that project to the hindbrain) represent the major contributors to hindbrain leptin action remains unclear.
Leptin Regulation of Glycemic Control
Mice null for leptin or LepRb display impaired glucose and insulin homeostasis, and submaximal doses of leptin that are insufficient to induce weight loss in ob/ob mice rapidly normalize blood glucose levels, suggesting that leptin effects on weight and glycemic control are dissociable (94). Similarly, restoration of LepRb expression selectively in the ARC of LepRb-null mice results in only a mild abrogation of obesity and hyperphagia, but normalizes blood glucose levels and amelioarates insulinemia (95). Though the mechanism remains unclear, LepRb/POMC neurons of the ARC specifically contribute to glucose homeostasis (96;97) The adjacent VMH is also rich with glucose-sensing neurons and may also participate in the regulation of glucose homeostasis (98). Hypothalamic LepRb-expressing glucose responsive neurons send relays via the autonomic nervous system that innervate the liver to mediate changes in glucose production and release (99). PI3-K plays a central role in regulating hepatic glucose flux (55).
Leptin Action in Reproduction
Leptin levels communicate whether there is sufficient energy available to undertake energy-demanding physiological functions, including reproduction. Accordingly, leptin restores reproduction to otherwise infertile leptin-deficient ob/ob mice (100). Leptin regulates the reproductive system via gonadotropinreleasing hormone (GnRH)-secreting neurons in the preoptic area, and these neurons in turn facilitate release of pituitary gonadotropins into the circulation. GnRH neurons do not express LepRb, however, indicating that leptin must act indirectly/trans-synaptically to regulate GnRH secretion (101). Several LepRb-expressing regions project to GnRH neurons and play roles in reproduction, including the ARC, medial preoptic area (MPOA) and the PMv. Leptin regulates expression of hypothalamic kisspeptin-1 (Kiss1) and galanin-like peptide (GALP), both of which regulate GnRH secretion, but it is unclear whether Kiss1 and GALP neurons co-express LepRb or are indirectly modulated by LepRb neurons (100;102). CART is also a facilitator of GnRH expression, and the large population of CART neurons in the ARC and PMv that project onto GnRH neuron-containing regions are potential sites of direct or indirect leptin regulation (103).
Leptin Regulation of Thyroid Function
Leptin regulates expression of the hypothalamic peptide thyrotropin-releasing hormone (TRH), which is crucial for production of thyroid hormones that contribute to energy metabolism (5). Fasting (i.e. low circulating leptin), suppresses TRH expression coordinately decreasing thyroid hormone levels and thyroid-mediated actions on metabolic rate. Leptin differentially regulates two subpopulations of TRH-expressing neurons of the PVN: one co-expresses TRH and LepRb (i.e. direct leptin regulation); the other population expresses melanocortin receptors and is regulated by ARC LepRb neurons (indirect leptin regulation) (104). While the function of the TRH/LepRb neurons remains unclear, the population of TRH neurons indirectly regulated by leptin feed into the hypothalamic-pituitary axis to regulate thyroid hormone secretion.
Leptin Regulation of the Mesolimbic Dopamine System
The incentive salience of palatable food can prompt ingestion irrespective of satiety signals (105;106). The most obvious analogy is that often we are full after eating a meal, but still eat dessert because it looks appealing and tasty. Leptin regulates the incentive value of food (as well as of other addictive substances, such as drugs of abuse) (105;107;108). Some of the neural mechanisms by which leptin may control food reward are beginning to be elucidated via the investigation of the interaction of leptin with the mesolimbic dopamine (DA) system (109–112). The mesolimbic DA system is composed of a set of DA neurons in the ventral tegmental area (VTA) that project forward to innervate the striatum, amygdala, and prefrontal cortex. This system is the locus of action for the reinforcing effects of drugs of abuse, and is also important to mediate the incentive salience of food and other natural rewards (106;113).
ARC LepRb neurons do not project to the VTA, but systemic leptin administration modulates food reward, suggesting a different locus for the regulation of DA signaling (105). A number of groups have reported the presence of LepRb-expressing VTA DA neurons and demonstrated the ability of leptin to alter the physiology of the DA system (109–112). Leptin activates VTA LepRb neurons and regulates DA synthesis and release to downstream mesolimbic system components (110;114;115).
In addition to LepRb neurons in the VTA, leptin-sensitive neurons in the lateral hypothalamic area (LHA) interact extensively with the mesolimbic DA system to regulate motivation and reward, including food reward (106;108;116). LHA orexin (OX) neurons project to the VTA and regulate drug and food-associated reward signaling; leptin inhibits activity of OX neurons, which could underly leptin suppression of hedonic reward signaling (116;117). Leptin also inhibits expression of the LHA neuropeptide MCH, and thus may inhibit the MCH neurons projecting to the NAc. Inhibition of OX and MCH signaling via their respective receptor antagonists inhibit food intake and modulates DA signaling in rodent models similar to leptin, and are thus being studied as potential drugs for anxiety and weight loss. We have also identified a novel population of LepRbexpressing neurons in the LHA that project to the VTA to regulate the mesolimbic DA system (our unpublished data). Thus, leptin acts via multiple ARC-independent systems to control the VTA and the mesolimbic DA system at its inception in the VTA, and these sites of leptin action likely regulate the incentive salience of food.
Summary
Leptin regulates a variety of diverse processes, from satiety to the incentive salience of food, and from reproduction to autonomic nervous system activity. While some aspects of how leptin signaling controls these diverse function remain to be established, it is clear that specific LepRb signals control specific physiologic functions. In order to understand how leptin and specific LepRb signals modulate all of its physiologic effects, it will be necessary to understand the neural and physiologic function of all LepRb neurons in the CNS, and to define how individual leptin signals contribute to the function of each population of neurons. Clearly, the field has a great deal of work to do over the next years.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Friedman JM, Halaas JL. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 2.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. J Comp Neurol. 2005;493:63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 3.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 4.Bates SH, Myers MG., Jr Trends Endocrinol.Metab. 2003;14:447–452. doi: 10.1016/j.tem.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Ahima RS, Prabakaran D, Mantzoros CS, Qu D, Lowell BB, Maratos-Flier E, Flier JS. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 6.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 7.Elmquist JK, Maratos-Flier E, Saper CB, Flier JS. Nature Neuroscience. 1998;1:445–449. doi: 10.1038/2164. [DOI] [PubMed] [Google Scholar]
- 8.Montague CT, Farooqi IS, Whitehead JP, Soos MS, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Early AR, Barnett AH, Prins JB, O'Rahilly S. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 9.Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, leBouc Y, Froguel P, Guy-Grand B. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 10.Zhang F, Basinski MB, Beals JM, Briggs SL, Clawson DK, DiMarchi RD, Furman TC, Hale JE, Hsiung HM, Schoner BE, Smith DP, Zhang XY, Wery JP, Shevitz RW. Nature. 1997;387:209. doi: 10.1038/387206a0. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 12.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 13.Uotani S, Bjorbaek C, Tornoe J, Flier JS. Diabetes. 1999;48:279–286. doi: 10.2337/diabetes.48.2.279. [DOI] [PubMed] [Google Scholar]
- 14.Gavrilova O, Barr V, Marcus-Samuels B, Reitman M. J.Biol.Chem. 1997;272:30546–30551. doi: 10.1074/jbc.272.48.30546. [DOI] [PubMed] [Google Scholar]
- 15.Yang G, Ge H, Boucher A, Yu X, Li C. Mol Endocrinol. 2004;18:1354–1362. doi: 10.1210/me.2004-0027. [DOI] [PubMed] [Google Scholar]
- 16.Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr J Biol Chem. 2002;277:41547–41555. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 17.Bahrenberg G, Behrmann I, Barthel A, Hekerman P, Heinrich PC, Joost HG, Becker W. Mol.Endocrinol. 2002;16:859–872. doi: 10.1210/mend.16.4.0800. [DOI] [PubMed] [Google Scholar]
- 18.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Mol.Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatti K, Farrar WL, Duhe RJ. Biochemistry. 2004;43:4272–4283. doi: 10.1021/bi036109b. [DOI] [PubMed] [Google Scholar]
- 20.White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA. J.Biol.Chem. 1997;272:4065–4071. doi: 10.1074/jbc.272.7.4065. [DOI] [PubMed] [Google Scholar]
- 21.Peelman F, Iserentant H, De Smet AS, Vandekerckhove J, Zabeau L, Tavernier J. J Biol Chem. 2006;281:15496–15504. doi: 10.1074/jbc.M512622200. [DOI] [PubMed] [Google Scholar]
- 22.Zabeau L, Defeau D, Van der HJ, Iserentant H, Vandekerckhove J, Tavernier J. Mol Endocrinol. 2004;18:150–161. doi: 10.1210/me.2003-0078. [DOI] [PubMed] [Google Scholar]
- 23.Matsuda T, Feng J, Witthuhn BA, Sekine Y, Ihle JN. Biochem.Biophys.Res.Commun. 2004;325:586–594. doi: 10.1016/j.bbrc.2004.10.071. [DOI] [PubMed] [Google Scholar]
- 24.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A. EMBO J. 1999;18:1309–1320. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, Sasaki M, Johnston JA, Yoshimura A. Genes Cells. 1999;4:339–351. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- 26.Kurzer JH, Argetsinger LS, Zhou YJ, Kouadio JL, O'Shea JJ, Carter-Su C. Mol Cell Biol. 2004;24:4557–4570. doi: 10.1128/MCB.24.10.4557-4570.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argetsinger LS, Kouadio JL, Steen H, Stensballe A, Jensen ON, Carter-Su C. Mol Cell Biol. 2004;24:4955–4967. doi: 10.1128/MCB.24.11.4955-4967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feener EP, Rosario F, Dunn SL, Stancheva Z, Myers MG., Jr Mol Cell Biol. 2004;24:4968–4978. doi: 10.1128/MCB.24.11.4968-4978.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishida-Takahashi R, Rosario F, Gong Y, Kopp K, Stancheva Z, Chen X, Feener EP, Myers MG., Jr Mol Cell Biol. 2006;26:4063–4073. doi: 10.1128/MCB.01589-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazurkiewicz-Munoz AM, Argetsinger LS, Kouadio JL, Stensballe A, Jensen ON, Cline JM, Carter-Su C. Mol Cell Biol. 2006;26:4052–4062. doi: 10.1128/MCB.01591-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG., Jr J Biol Chem. 2007;282:31019–31027. doi: 10.1074/jbc.M702838200. [DOI] [PubMed] [Google Scholar]
- 32.Schindler C, Darnell JE., Jr Annu.Rev.Biochem. 1995;64 doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 33.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 34.Mutze J, Roth J, Gerstberger R, Hubschle T. Neurosci.Lett. 2007;417:286–291. doi: 10.1016/j.neulet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- 35.De Souza D, Fabri LJ, Nash A, Hilton DJ, Nicola NA, Baca M. Biochemistry. 2002;41:9229–9236. doi: 10.1021/bi0259507. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Friedman JM. Proc Natl Acad Sci U.S.A. 1999;96:9677–9682. doi: 10.1073/pnas.96.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carpenter LR, Farruggella TJ, Symes A, Karow ML, Yancopoulos G. Proc Natl Acad Sci U S A. 1998;95:6061–6066. doi: 10.1073/pnas.95.11.6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG, Jr, Flier JS. J Biol Chem. 2001;276:4747–4755. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- 39.Banks AS, Davis SM, Bates SH, Myers MG., Jr J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 40.Bjorbaek C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 41.Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjorbaek C, Myers MG., Jr J Clin.Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn SL, Bjornholm M, Bates SH, Chen Z, Seifert m, Myers MG., Jr Mol Endocrinol. 2005;19:925–938. doi: 10.1210/me.2004-0353. [DOI] [PubMed] [Google Scholar]
- 43.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Nat.Med. 2004 doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 44.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Nat.Med. 2004 doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 45.Zhang EE, Chapeau E, Hagihara K, Feng GS. Proc.Natl.Acad.Sci.U.S.A. 2004;101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Molecular Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 47.Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier J, Joost HG, Becker W. FEBS J. 2005;272:109–119. doi: 10.1111/j.1742-4658.2004.04391.x. [DOI] [PubMed] [Google Scholar]
- 48.Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA. Proc.Natl.Acad.Sci.U.S.A. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bates SH, Stearns WH, Schubert M, Tso AWK, Wang Y, Banks AS, Dundon TA, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 50.Bates SH, Kulkarni RN, Seifert m, Myers MG., Jr Cell Metabolism. 2005;1:169–178. doi: 10.1016/j.cmet.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Bates SH, Dundon TA, Seifert m, Carlson M, Maratos-Flier E, Myers MG., Jr Diabetes. 2004;53:3067–3073. doi: 10.2337/diabetes.53.12.3067. [DOI] [PubMed] [Google Scholar]
- 52.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 53.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 54.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 55.Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 56.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ, Vanhaesebroeck B. Nature. 2006;441:366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 57.Duan C, Li M, Rui L. J.Biol Chem. 2004;279:43684–43691. doi: 10.1074/jbc.M408495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ren D, Li M, Duan C, Rui L. Cell Metab. 2005;2:95–104. doi: 10.1016/j.cmet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 59.Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L. J Clin.Invest. 2007;117:397–406. doi: 10.1172/JCI29417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr J Clin.Invest. 2005;115:3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McMinn JE, Liu SM, Liu H, Dragatsis I, Dietrich P, Ludwig T, Boozer CN, Chua SC., Jr Am.J Physiol Endocrinol.Metab. 2005;289:E403–E411. doi: 10.1152/ajpendo.00535.2004. [DOI] [PubMed] [Google Scholar]
- 62.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elmquist JK, Elias CF, Saper CB. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 64.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- 65.Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D, Jr, Breininger JF, Jonak Z, Schaefer J, Krouse M, Burghardt C, Campfield LA, Burn P, Kochan JP. J Histochem.Cytochem. 1999;47:353–362. doi: 10.1177/002215549904700309. [DOI] [PubMed] [Google Scholar]
- 66.Leshan RL, Bjornholm M, Munzberg H, Myers MG., Jr Obesity.(Silver.Spring) 2006;14 Suppl 5:208S–212S. doi: 10.1038/oby.2006.310. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 68.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 69.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 70.Butler AA, Cone RD. Neuropeptides. 2002;36:77–84. doi: 10.1054/npep.2002.0890. [DOI] [PubMed] [Google Scholar]
- 71.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Nat.Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 72.Ste ML, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. Proc.Natl.Acad.Sci.U.S.A. 2000;97:12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 74.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van Der Ploeg LH. Nat.Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 75.Accili D, Arden KC. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 76.Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- 77.Bousquet C, Ray DW, Melmed S. J Biol Chem. 1997;272:10551–10557. doi: 10.1074/jbc.272.16.10551. [DOI] [PubMed] [Google Scholar]
- 78.Kitamura T, Feng Y, Ido KY, Chua SC, Xu AW, Barsh GS, Rossetti L, Accili D. Nat.Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 79.Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Am.J Physiol Endocrinol.Metab. 2005;289:E1051–E1057. doi: 10.1152/ajpendo.00094.2005. [DOI] [PubMed] [Google Scholar]
- 80.MacNeil DJ, Howard AD, Guan X, Fong TM, Nargund RP, Bednarek MA, Goulet MT, Weinberg DH, Strack AM, Marsh DJ, Chen HY, Shen CP, Chen AS, Rosenblum CI, MacNeil T, Tota M, MacIntyre ED, Van Der Ploeg LH. Eur.J.Pharmacol. 2002;450:93–109. doi: 10.1016/s0014-2999(02)01989-1. [DOI] [PubMed] [Google Scholar]
- 81.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 82.Morton GJ, Niswender KD, Rhodes CJ, Myers MG, Jr, Blevins JT, Baskin DG, Schwartz MW. Endocrinology. 2003;144:2016–2024. doi: 10.1210/en.2002-0115. [DOI] [PubMed] [Google Scholar]
- 83.van de WE, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, Elmquist J, Lowell BB, Barsh GS, de Luca C, Myers MG, Jr, Schwartz GJ, Chua SC., Jr Endocrinology. 2007 doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaelin CB, Gong L, Xu AW, Yao F, Hockman K, Morton GJ, Schwartz MW, Barsh GS, MacKenzie RG. Mol Endocrinol. 2006;20:2591–2602. doi: 10.1210/me.2006-0107. [DOI] [PubMed] [Google Scholar]
- 85.Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Endocrinology. 2006;148:72–80. doi: 10.1210/en.2006-1119. [DOI] [PubMed] [Google Scholar]
- 86.Piper ML, Unger EK, Myers MG, Jr, Xu AW. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sternson SM, Shepherd GM, Friedman JM. Nat.Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- 88.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 89.Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S, Jr, Elmquist JK, Lowell BB. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 90.Grill HJ. Obesity.(Silver.Spring) 2006;14 Suppl 5:216S–221S. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- 91.Huo L, Maeng L, Bjorbaek C, Grill HJ. Endocrinology. 2007;148:2189–2197. doi: 10.1210/en.2006-1572. [DOI] [PubMed] [Google Scholar]
- 92.Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW. J.Clin.Invest. 2005;115:703–710. doi: 10.1172/JCI200522081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams DL, Baskin DG, Schwartz MW. Diabetes. 2006;55:3387–3393. doi: 10.2337/db06-0558. [DOI] [PubMed] [Google Scholar]
- 94.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 95.Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Jr, Lowell BB, Elmquist JK. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 96.Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Cell Metab. 2006;4:123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 97.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. J Clin.Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oomura Y. New Physiol.Sci. 1987;2:199–203. [Google Scholar]
- 99.Pocai A, Obici S, Schwartz GJ, Rossetti L. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Tena-Sempere M. Neuroendocrinology. 2006;83:275–281. doi: 10.1159/000095549. [DOI] [PubMed] [Google Scholar]
- 101.Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL. Neuroendocrinology. 1998;67:370–376. doi: 10.1159/000054335. [DOI] [PubMed] [Google Scholar]
- 102.Dungan HM, Clifton DK, Steiner RA. Endocrinology. 2006;147:1154–1158. doi: 10.1210/en.2005-1282. [DOI] [PubMed] [Google Scholar]
- 103.Rondini TA, Baddini SP, Sousa LF, Bittencourt JC, Elias C. Neuroscience. 2004;125:735–748. doi: 10.1016/j.neuroscience.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 104.Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN. J Clin Invest. 2001;107:111–120. doi: 10.1172/JCI10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Figlewicz DP, Naleid AM, Sipols AJ. Physiol Behav. 2006 doi: 10.1016/j.physbeh.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kelley AE, Berridge KC. J Neurosci. 2002;22:3306–3311. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carr KD. Physiol Behav. 2006 [Google Scholar]
- 108.Fulton S, Richard D, Woodside B, Shizgal P. Behav.Brain Res. 2004;155:319–329. doi: 10.1016/j.bbr.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 109.Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 110.Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 111.Figlewicz DP. Am.J Physiol Regul.Integr.Comp Physiol. 2003;284:R882–R892. doi: 10.1152/ajpregu.00602.2002. [DOI] [PubMed] [Google Scholar]
- 112.Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Brain Res. 2003;964:107–115. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- 113.Nestler EJ. Nat.Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- 114.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, Noguera I, Torres R, Macdonald LE, Stewart AF, DeChiara TM, Yancopoulos GD. Nat.Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 115.Roseberry AG, Painter T, Mark GP, Williams JT. J Neurosci. 2007;27:7021–7027. doi: 10.1523/JNEUROSCI.1235-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.DiLeone RJ, Georgescu D, Nestler EJ. Life Sci. 2003;73:759–768. doi: 10.1016/s0024-3205(03)00408-9. [DOI] [PubMed] [Google Scholar]
- 117.Harris GC, Wimmer M, Aston-Jones G. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]