

Abstract
The enzyme tRNA(m1G37) methyl transferase catalyzes the transfer of a methyl group from S-adenosyl methionine (AdoMet) to the N1 position of G37, which is 3′ to the anticodon sequence and whose modification is important for maintaining the reading frame fidelity. While the enzyme in bacteria is highly conserved and is encoded by the trmD gene, recent studies show that the counterpart of this enzyme in archaea and eukarya, encoded by the trm5 gene, is unrelated to trmD both in sequence and in structure. To further test this prediction, we seek to identify residues in the second class of tRNA(m1G37) methyl transferase that are required for catalysis. Such residues should provide mechanistic insights into the distinct structural origins of the two classes. Using the Trm5 enzyme of the archaeon Methanocaldococcus jannaschii (previously MJ0883) as an example, we have created mutants to test many conserved residues for their catalytic potential and substrate-binding capabilities with respect to both AdoMet and tRNA. We identified that the proline at position 267 (P267) is a critical residue for catalysis, because substitution of this residue severely decreases kcat of the methylation reaction in steady-state kinetic analysis, and kchem in single turnover kinetic analysis. However, substitution of P267 has milder effect on Km and little effect on Kd of either substrate. Because P267 has no functional side chain that can directly participate in the chemistry of methyl transfer, we suggest that its role in catalysis is to stabilize conformations of enzyme and substrates for proper alignment of reactive groups at the enzyme active site. Sequence analysis shows that P267 is embedded in a peptide motif that is conserved among the Trm5 family, but absent from the TrmD family, supporting the notion that the two families are descendants of unrelated protein structures.
The discovery that proteins of unrelated structures can catalyze the same enzymatic activity has important implications for the evolution of biological functions. Such unrelated proteins provide insights into nature’s capacity of creating different solutions to thermodynamic problems of the same biological reactions. Particularly significant is when unrelated proteins for the same reaction are found in different domains of life. Such events are known as non-orthologous gene displacement (1-3), which are frequently observed for DNA replication proteins, such as primases, helicases, ligases, and the repair DNA polymerases (2). Recent studies are beginning to uncover non-orthologous gene displacement for proteins that recognize RNA. For example, there are two distinct structural classes of lysyl-tRNA synthetases (LysRS) that catalyze aminoacylation of tRNA with lysine (4): the class I LysRS enzymes are prevalent in archaea, whereas class II enzymes are in bacteria and eukarya. The CCA-adding enzymes that catalyze addition of CCA to the tRNA 3′ end are also divided into two structural classes (5): class I enzymes reside in archaea, whereas class II enzymes reside in bacteria and eukarya. The widespread occurrence of such examples in protein enzymes that recognize DNA and RNA suggests that non-orthologous gene displacement is an important process in the history of decoding genetic information.
We are interested in the enzyme tRNA(m1G37) methyl transferase that catalyzes transfer of a methyl group from S-adenosyl methionine (AdoMet) to the N1 position of G37 (6). The m1G37 modification is located 3′ to the anticodon in tRNAs specific for leucine (CUN codons, N being any of the four natural nucleotides), proline (CCN), one of the arginine tRNAs (CGG), and often in tRNAs that contain G37. This modification is conserved in all three domains of life for the aforementioned tRNAs, and is also present in mitochondria and chloroplasts (7). The presence of m1G37 modification has been shown to prevent frame-shifts during decoding of genetic information (7-9). Earlier genetic studies had led to the identification of the enzyme tRNA(m1G37) methyl transferase as encoded by the trmD gene in bacteria (6), and more recently the trm5 gene in yeast (10). Deletion or inactivation of trmD or trm5 has led to severe growth defect in certain bacteria and death in others (10, 11). Interestingly, the archaeal counterpart of trmD or trm5 remained elusive, until a genetic screen identified a gene of Methanocaldococcus vannielii that rescued a temperature-sensitive trmD mutant of bacteria (10). The homolog of the M. vannielii gene in M. jannaschii is Mj0883, which we have now shown as the gene that encodes the enzyme tRNA(m1G37) methyl transferase (12). Interestingly, extensive sequence and bioinformatic analysis had previously suggested that the gene product of Mj0883 would be related to those of the eukaryotic trm5 but would be distinct from those of trmD genes, such that the two proteins could be considered as descendants of unrelated families (13, 14). Our recent biochemical studies support this division, and suggest that Mj0883 and trm5 genes can be grouped into one class while trmD genes into another (12). Because of this classification, we have now renamed Mj0883 as the trm5 gene of M. jannaschii.
The separation of trmD and trm5 into two classes is based on the following criteria. First, members of the proteins TrmD and Trm5, while conserved within their own class, do not share sequence similarity between the two classes (12-14). There have been three X-ray crystal structures of TrmD in recent years (15-17). These structures reveals that the TrmD enzyme is a homo-dimer, and that the highly conserved catalytic core is built by the dimer interface that binds the methyl donor AdoMet. In contrast, biochemical studies suggest that M. jannaschii Trm5 is a monomer, and that conserved residues that are likely to bind AdoMet are distinct from those of TrmD (12). The monomer structure has also been reported for the human Trm5 (18). Second, the AdoMet substrate in each subunit of the TrmD crystal structure is recognized by a trefoil knot structure (15-17), where the polypeptide chain threads through a deeply twisted loop and tucks many residues (> 15) through the loop. In the available protein data base of structural motifs that bind AdoMet, the trefoil knot structure is of the class IV fold (19), which is rare and is only observed in the SPOUT (SpoU-TrmD) enzyme family (14, 20). However, although no crystal structures exists for Trm5 at present, modeling and bioinformatics suggests that Trm5 would recognize AdoMet using the class I fold, which is one of the most common structural folds among the co-factor-dependent methyl transferases (12-14). This structural fold lacks the knotted motif, but contains an open sandwich-like domain that is found in the ancient Rossmann-fold for nucleotide-binding (21). Sequence and structural modeling of Trm5 (13, 14), together with mutational analysis (12), has established that Trm5 is evolutionarily distinct from TrmD.
To gain more insights into the relationship between the two classes of tRNA(m1G37) methyl transferase, it will be necessary to elucidate and compare the catalytic motifs of each class. In the case of TrmD, the available crystal structures allow some insights into the catalytic strategy of the enzyme, although no structural information is available regarding binding of the tRNA substrate. In the case of M. jannaschii Trm5, even less is known. Of interest are the similarities and potential differences of the archaeal Trm5 from other class I methyl transferases. A relevant example is the relationship between the RNA:m5C methyl transferase and DNA:m5C methyl transferase. Although these m5C methyl transferase enzymes have the same class I AdoMet-binding fold and catalyze the same reaction, they use different sequence motifs for catalysis (22, 23). This example illustrates the possibility of sequence permutations among the class I family of methyl transferases leading to different arrangements of catalytic motifs (24). To address the deficiency in our general knowledge of archaeal Trm5, we present below a series of experiments to search for residues in Trm5 that are important for catalysis. In our previous study, using the M. jannaschii Trm5 as an example (12), we have surveyed a few conserved motifs by mutagenesis and identified a conserved proline that is critical for the activity of the enzyme. However, many other conserved motifs were not tested, raising the question of whether any of the untested residues could be important for activity. Additionally, of residues that are important for activity (such as the aforementioned proline), we wish to determine if they contribute to catalysis or to substrate binding. In this study, we have developed new tools to evaluate substrate binding and catalytic competence of the wild-type and mutants of M. jannaschii Trm5. In particular, we have established conditions of transient-state kinetics that enables us to assess catalysis in the active site of the enzyme. Results of these studies, together with those based on steady-state kinetic analysis, represent an important step forward to understand the catalytic mechanism of an archaeal Trm5.
Experimental Procedures
Enzyme purification and mutagenesis
An over-expression clone of the Trm5 of M. jannaschii, containing a C-terminal His tag, was constructed in the pET-22b vector and expressed and purified from E. coli BL21(DE3) as described (12). Site-directed mutations in the enzyme were created by the QuikChange protocol and confirmed by sequence analysis. Variants of enzymes were purified by the same procedure as the wild-type.
Enzyme kinetic assays
Conditions for the methylation assay in steady-state kinetics at 50 °C were as described in 0.1 M Tris-HCl, pH 8.0, 4 mM DTT, 0.1 mM EDTA, 6 mM MgCl2, 0.1 M KCl, 0.024 mg/mL BSA, using 50 μM [3H] AdoMet (0.02 mCi/mL, 870 dpm/pmole, Sigma) as the cofactor, and the transcript of M. jannaschii tRNACys (0.5-10 μM) as the substrate (12). Enzyme assays were performed with time courses in the linear phase of product formation. Steady-state parameters of the AdoMet or tRNA substrate were determined from plots of different substrate concentrations versus velocities, where substrate concentrations ranged in 0.2-5.0-fold of Km. The data were fit to the Michaelis-Menten equation, using the KaleidaGraph software (Synergy software, version 3.6). Transient-state kinetics were performed on a rapid quench RQF-3 KinTek instrument, using the constant quench option. The standard buffer was the same as that used in steady-state analysis, while the quench buffer contained 2.5 M sodium acetate, pH 5.0. Reaction aliquots of 30 μL (15 μL each syringe) were stopped with 55 μL of the quench solution, and 20 μL was precipitated on filter pads by 5% trchloroacetic acid. The acid precipitable counts were measured by the Beckman liquid scintillation counter LS6000SC.
Determination of tRNA binding by gel shift
The T7 transcript of M. jannaschii tRNACys was labeled by the CCA enzyme, using [α-32P]ATP in an exchange reaction, and purified by a denaturing PAGE/7 M urea. The labeled tRNA (5,000 cpm, 5 nM) was mixed with increasing concentrations of the WT or a variant Trm5 in a binding buffer (25 mM sodium phosphate (pH 6.8), 50 mM KCl, 10 mM MgCl2) at room temperature for 15 min, adjusted with an equal volume of a native loading dye (0.1% each xylene cyanol and bromophenol blue in the binding buffer), cooled at 4 °C for 15 min, and separated by a 2% native agarose gel in 0.5 × TBE for 2.5 hrs, 4 °C at 34 volts. The gel was dried and developed by a phosphorimager (Molecular Dynamics), where the fraction of bound and free tRNA in each binding reaction were quantified, using Image Quant. The data were fit to a hyperbola to derive the Kd of the binding interaction.
Determination of AdoMet-binding by fluorescence and by cross-linking
Titrations of AdoMet by steady-state fluorescence were performed on a PTI (Photon Technology International) bench top continuous spectro-fluorometer model 814 (Lawrenceville, NJ). Fluorescence emission spectra were obtained by exciting Trm5 (1 μM) at 282 nm and scanning from 300-400 nm at 1.0 nm increments. The excitation and emission monochromators were set at slit widths of 0.5 mm. The photomultiplier voltage was set at 1100 V, and data were collected using the FeliX32 software and imported into Excel and Sigma plot for graphic presentation. Photo-bleaching was determined to be minimal under the time scale of these experiments, by using a stir bar during scanning and by ensuring that repetitive measurements yielded the same fluorescence emission intensity. After obtaining the emission scan for the enzyme alone (0.1 M Tris-HCl, pH 7.5, 100 mM KCl, 6 mM MgCl2, 4 mM DTT, and 0.1 mM EDTA), which yielded 5 × 105 counts/sec, AdoMet was added at the indicated concentrations (0.4-113 μM), mixed, allowed to bind for 5 min at 25 °C, and the emission scan observed from 300-400 nm. The reactions were held at 25 °C during the scan time (approximately 1 min). The changes of intensity at 340 nm versus AdoMet concentrations were fit to a hyperbola equation to derive the Kd for AdoMet.
The cross-linking assay was performed by incubating M. jannaschii Trm5 (16 μM) with 2.5 μM of [methyl-3H]AdoMet (0.5 μCi, 78 Ci/mmol, NEN) overnight at 4 °C. The binding reactions were added to a 96-well plate on ice and placed 8 cm from a UV source at 312 nm for 20 min at 4 °C. The reactions were then separated by SDS-PAGE, stained with Coomassie blue, followed by incubation with En3hance (Perkin Elmer 6NE9701) for 30 min. The gel was then dried, exposed to a tritium screen (Amersham Biosciences) for one week, and subjected to fluorographic analysis, using Image Quant (Molecular Dynamics).
Results
Mutational analysis of M. jannaschii Trm5
An extensive sequence alignment of M. jannaschii Trm5 and its archaeal and eukaryotic homologs was established (Fig.1). To better interpret the sequence alignment, we performed homology modeling by using the available crystal structure of MJ0882 ORF (25) as a reference. We have previously shown that the sequence of MJ0882 was the closest homolog of the sequence of M. jannaschii Trm5 in the entire protein data bank (PDB) of AdoMet-dependent methyl transferases (12). The gene of MJ0882 is located immediately 5′ to the gene of M. jannaschii Trm5 (previously MJ0883). The protein sequence of MJ0882 is homologous to RsmC (the methyl transferase for the m2G1207 methylation of 16S rRNA (26)) and RsmD of bacteria. The crystal structure of MJ0882 was solved as part of the Structural Genomics Initiatives (PDB code 1DUS; (25)). Using the structure of MJ0882 as a homology model, we showed that Trm5 is divided into three domains: an N- and C-terminal domain (Fig.1A,C) and a middle domain that has the AdoMet-binding motifs (Fig.1B). Characteristic secondary structural elements in the AdoMet domain that are predicted to bind the cofactor are inferred from the structure of MJ0882 (Fig.1D).
Fig.1.

Sequence alignment of archaea and eukaryotic Trm5 proteins by ClustalW, together with modeling of the alignment based on the crystal structure of MJ0882 (25). Significance scores are not calculated for this alignment. Sequences used in the alignment include Trm 5 of M. jannaschii (Mj), Methanobacterium thermoautotrophicum (Mth), M. acetivorans (Ma), M. mazei (Mz), Halobacterium halobium (Hh), Neurospora crassa (Nc), Arabidopsis thalania (At), Drosophila melanogaster (Dm), Anopheles gambiae (Ag), Homo sapiens (Hs), and Saccharomyces cerevisiae (Sc). Characteristic secondary structural elements α-helices and β-strands are indicated by arrows and cylinders, respectively, across the top of the alignment. Mutations that had been previously published are indicated by circles, while those that were created in this study are indicated by vertical arrows. Trm 5 is divided into three domains: (A) the N-terminal domain, (B) the AdoMet-binding domain, and (C) the C-terminal domain. In (D), crystal structure of MJ0882 is shown, where cylinders and arrows represent α-helices and β-strands, respectively, according to the published assignments in the crystal structure (25). No additional modeling is performed on the structure of MJ0882, thus no ProsaII or Verify3D is employed. The positions of G205, G207, N265, and P267 residues of M. jannaschii Trm5 are deduced from the sequence alignment with MJ0882, and are shown in red, indicating their positions in the AdoMet-binding site.
Conserved residues in the AdoMet domain have been noted previously (12). Six of these conserved residues had been tested by alanine substitutions, which included D201 at the end of the β1 strand, D223 at the end of the β2 strand (the D loop region (25)), N265 and P267 of the NLP motif at the end of the β4 strand (the P loop region (25)), and the two glycines in the FAGxG motif (the G loop region (25)) which is known for binding the methionine moiety of AdoMet (27). While mutants harboring one or two of these substitutions lost the methylase activity to varying degrees, the P267A mutant had the most severe defect, exhibiting a loss of ∼100-fold in activity under the previous assay condition (12). However, because there remained many other conserved residues that were not tested, it was not known whether the P267A defect was indeed the most severe.
To identify the catalytic motif in M. jannaschii Trm5, the entire protein sequence was searched for residues that were strictly or highly conserved among the Trm5 family (Fig.1). A total of 18 residues were identified, including 3 in the N-terminal domain (D109, R144, E149), 3 in the C-terminal domain (V317, K318, P322), and the remaining 12 in the AdoMet domain (Y176, G205/G207, D223, N225, P226, N239, D251. I263, M264, N265, L266, and P267). Variants were created to harbor a single substitution with alanine, except for one that contained double substitutions of G205/G207 with alanine. All variants were expressed in E. coli, and were purified using the procedure established for the wild-type, including a step of heat-denaturation (due to the thermophilic nature of M. jannaschii). The resistance to high temperatures suggests that all of these mutants maintained the inherent thermo-stability of the wild-type. These variants were tested for the tRNA(m1G37) methyl transferase activity, using [3H]-AdoMet as the cofactor and the unmodified transcript of M. jannaschii tRNACys as the substrate, which contains G37 and can be modified (12). For the purpose of rapidly screening the 18 mutants, the activity was assayed under single turnover conditions, where the concentration of enzyme was 3-5 fold in excess of the tRNA concentration (at 1 μM). This analysis showed that the majority of these variants were active, displaying plateau levels of methylation within 5-fold of that of the wild-type (data not shown). However, 5 variants had a more reduced plateau, which included the R144A mutant containing a substitution in the N-terminal domain, and Y176A, G205/G207A, D223A, and P267A mutants containing substitutions in the AdoMet domain. These 5 variants were further analyzed (see below). No mutations in the C-terminal domain were found to be defective.
Steady-state kinetic analysis was performed previously only on the wild-type enzyme. Thus, to determine the defect of the 5 variants, they were analyzed similarly to derive their Km and kcat parameters for the tRNA substrate, and to compare their parameters with those of the wild-type enzyme (Table 1). Under saturating concentrations of the AdoMet cofactor but varying tRNA concentrations, time courses were taken in the linear phase of product formation and kinetic parameters were determined from plots of velocities versus tRNA concentrations that ranged from 0.2 to 5 times of Km. The data were fit to the Michaelis-Menten equation, using the KaleidaGraph software. Compared to the wild-type (kcat =0.5 min-1, and Km = 0.73 μM), all 5 variants showed decreased kcat, with the largest decrease of nearly 100-fold exhibited by the P267A mutant (kcat = 0.006 min-1). All 5 variants also showed increased Km, with the largest increase of almost 10-fold by the P267A mutant (Km = 6.0 μM). Together, the P267A mutant harbored the largest defect in kcat/Km, about 1000-fold down from that of the wild-type. This effect is larger than the previously estimated ∼100-fold effect of the mutant, and it firmly established the importance of the P267 residue.
Table 1.
Kinetic parameters with respect to tRNA in the methyl transferase reaction
| kcat (min-1) | Km (μM) | kcat/Km (μM-1min-1) | Relative kcat/Km | ||
|---|---|---|---|---|---|
| Wild-type Trm5 | 0.5 ± 0.08 | 0.7 ± 0.03 | 0.7 ± 0.05 | 1.0 | |
| √ | R144A | 0.2 ± 0.1 | 4.3 ± 0.3 | 0.04 ± 0.2 | 0.06 |
| √ | Y176A | 0.08 ± 0.001 | 2.5 ± 0.01 | 0.03 ± 0.003 | 0.05 |
| √ | G205A/G207A | 0.09 ± 0.01 | 4.6 ± 1.5 | 0.02 ± 0.02 | 0.03 |
| √ | D223A | 0.02 ± 0.006 | 3.0 ± 0.6 | 0.008 ± 0.006 | 0.01 |
| N225A | 0.22 ± 0.02 | 7.2 ± 0.1 | 0.03 ± 0.02 | 0.05 | |
| P226A | 0.11 ± 0.01 | 3.0 ± 0.2 | 0.038 ± 0.04 | 0.06 | |
| N265A | 0.08 ± 0.02 | 8.3 ± 0.5 | 0.01 ± 0.01 | 0.01 | |
| √ | P267A | 0.006 ± 0.001 | 6.0 ± 0.9 | 0.001 ± 0.001 | 0.001 |
Kinetics of the wild-type Trm5 was assayed with 50 nM enzyme and tRNA concentrations ranging in 0.5-8.0 μM. Kinetics of mutants was assayed with 0.3-1.0 μM enzyme and tRNA ranging in 2.5-20 μM. All kcat and Km parameters were the average of at least 3 independent measurements. The √ marked the five mutants whose activities were below 20% of that of the wild-type in the initial screening experiments.
P267 is in the NLP motif of Trm5 that resembles the NPPY motif of DNA or RNA methyl transferases that target the exocyclic amino group of adenine, cytosine, or guanine for methylation. A relaxed form of the NPPY motif (N/D/S)-(I/P)-P-(Y/F/W/H) has also been reported for some methyl transferases (28). In the N6-adenine DNA methyl transferase M·TaqI, for example, the Asn and Pro residues of the NPPY motif (underlined) are both catalytically important (29). In particular, substitution of the Asn results in complete inactivation of the enzyme (30). However, because Trm5 modifies the cyclic N1, but not the exocyclic N6, it might potentially differ from DNA methyl tranferases by exploiting the NLP motif differently. To test this possibility, the asparagine mutant in the NLP motif (N265A) was examined by the detailed kinetic analysis with respect to tRNA. In addition, because a separate NP motif was identified at the end of the β4 strand (Fig.1B), the N225A and P226A substitutions were also analyzed. Although these three additional mutants were all defective in both kcat and Km with respect to tRNA (Table 1), their overall kcat/Km decreased 15- to 100-fold, smaller than the decrease observed of the P267A mutant. Thus, of the 18 variants that were examined, the P267A mutant indeed showed the most severe defect in the methyl transferase activity. These experiments also revealed that, unlike DNA methyl transferases, N265 in Trm5 makes less kinetic contribution than P267, even though both amino acids are in the same NLP motif.
The P267A mutant was then examined by steady-state analysis with respect to the AdoMet substrate (Table II) to determine its kinetic parameters with the cofactor. A series of initial rates was obtained by assaying varying concentrations of the cofactor with saturating concentrations of tRNA. Analysis of the relationship between rates and AdoMet concentrations showed that the wild-type kcat (0.73 min-1) and Km (1 μM) were similar to those published recently (12). However, the P267A mutant exhibited a small improvement in Km (0.5 μM), but a large decrease in kcat by ∼100-fold, leading to an overall loss of kcat/Km of 50-fold. Thus, compared to the larger tRNA substrate, the kcat defect of the P267A mutant is the same for the cofactor, confirming the catalytic deficiency of the mutant. The difference in the overall kcat/Km between the tRNA-dependent and factor-dependent reactions therefore resides in the Km effect. The greater Km effect with respect to the tRNA substrate suggests that the mutation specifically interferes with the steps that involve tRNA in the catalytic pathway, such as binding of tRNA and the proper alignment of G37 for methyl transfer.
Table 2.
Kinetic parameters with respect to AdoMet in the methyl transferase reaction
| kcat (min-1) | Km(μM) | kcat/Km (μM-1min-1) | Relative kcat/Km | |
|---|---|---|---|---|
| Wild-type Trm5 | 0.73 ± 0.01 | 1.0 ± 0.1 | 0.7 ± 0.1 | 1.0 |
| P267A | 0.006 ± 0.001 | 0.5 ± 0.04 | 0.01 ± 0.003 | 0.02 |
Kinetics of the wild-type Trm5 and P267A mutant was assayed with 0.2 μM enzyme, 6 μM tRNACys, and AdoMet ranging in 0.5-15 μM. The kcat and Km values are the average of at least 3 independent measurements.
Analysis of substrate binding
Because a detailed kinetic mechanism has not been established for tRNA(m1G37) methyl transferase, and it is not known if Km can be interpreted as the affinity for a substrate, we developed assays to determine if the P267A mutant was defective in the overall substrate binding affinity. The affinity for tRNA was tested by a gel shift assay for M. jannaschii Trm5, similar to that described previously (12), where the transcript of M. jannaschii tRNACys was labeled, and its mobility shift in the presence of increasing concentrations of enzyme was examined by electrophoresis on an agarose gel in native conditions. The amounts of free and bound tRNA in each binding reaction were quantified by a phosphor-imager and the fraction of bound was calculated and plotted against enzyme concentration. Because the tRNA concentration in the binding assay was low (5 nM) relative to the range of enzyme concentrations (0.5-16 μM) (e.g. Fig.2), the enzyme concentration that yielded 50% shift in the binding curve provided the value for an apparent Kd. This analysis revealed a Kd of the wild-type enzyme for tRNA at 0.4 μM (Table 3), which is in good agreement with the Kd obtained previously (12) and is similar to the Km of tRNA (Table 1).
Fig.2.
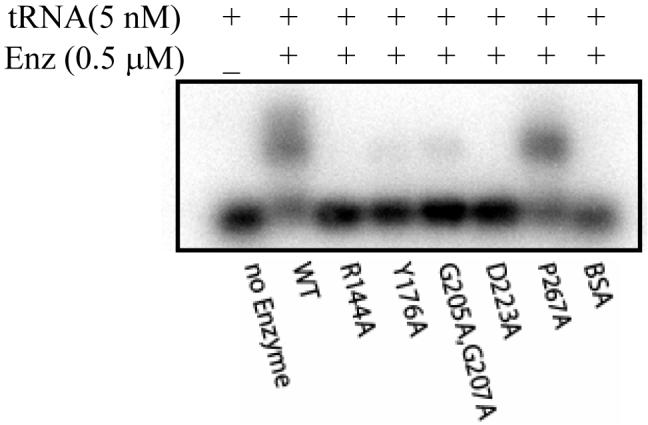
Gel shift analysis of tRNA-binding showing the roughly 50% mobility shift of tRNACys by the wild-type Trm5 and P267A mutant at 0.5 μM, but less than 10% shift by the Y176A and G205A/G207A mutants, and no shift by the R144A and D223A mutants.
Table 3.
Determination of the equilibrium dissociation constant (Kd) for tRNA by gel shift
| WT | R144A | Y176A | G205A/G207A | D223A | P267A | |
| Kd (μM) | 0.4 ± 0.1 | 3.9 ± 0.6 | 1.7 ± 0.1 | 1.0 ± 0.4 | 4.6 ± 0.8 | 0.3 ± 0.1 |
The 5 defective variants that displayed lower than 20% of the wild-type activity level (Table 1) were each assayed by the gel shift assay to determine their Kd for tRNA. Of the 5 variants, only the P267A mutant maintained the Kd at the wild-type level, whereas the other 4 mutants showed an elevated Kd, indicating reduced affinity for the tRNA substrate by 2-10 fold (Table 3). These affinities were then re-examined by assaying each of these mutants at a concentration that caused the wild-type enzyme to shift roughly 50% of the tRNA substrate (0.5 μM, Fig.2). This analysis confirmed that only the P267A mutant was able to shift the tRNA substrate similarly as the wild-type, whereas the other 4 mutants had a weaker (Y176A, G205A/G207A) or non-shifting (R144A, D223A) activity. The generic protein BSA served as a control to exclude the possibility of non-specific gel shift. Thus, based on the gel shift assay, the P267A mutant had the same binding capacity for the tRNA substrate as the wild-type enzyme. The lack of tRNA-binding by the R144A and D223A mutants suggests that the R144 and D223 residues may provide motifs for tRNA-binding.
A fluorescence titration assay was developed to measure the enzyme binding affinity for the AdoMet substrate. M. jannaschii Trm5 contains only one tryptophan (W169) in the β2 strand of the AdoMet domain (Fig.1B), which served as an intrinsic fluorescence probe. Excitation of the enzyme at 282 nm produced a typical protein emission spectrum between 300 and 400 nm (Fig.3A). Addition of AdoMet quenched the emission spectrum, with the largest quench at 340 nm. To ensure that the quench was specific to Trm5, a solution of free tryptophan was used as a control and its emission spectrum was monitored. Under the identified excitation (282 nm) and emission (340 nm) wavelengths, addition of AdoMet to the control yielded minimum quench (Fig.3A), confirming that the quench observed for Trm5 was specific to the enzyme. Addition of increasing concentrations of AdoMet to Trm5 progressively quenched the tryptophan fluorescence intensity, such that decreases of the intensity at 340 nm versus the cofactor concentrations could be fit to a hyperbolic equation to derive the Kd for the cofactor (Fig.3B). This analysis showed that the Kd of the wild-type for AdoMet was 3.2 μM, and that of the P267A mutant was 2-fold higher at 6.4 μM (Fig.3B). Although the fluorescence change upon binding the AdoMet ligand is reduced by 2-fold in the mutant, the overall change in Kd was small. Thus, while the mutation appeared to alter the binding interaction with AdoMet, as indicated by the overall reduced fluorescence change, it nonetheless does not cause a major loss of binding affinity for the cofactor.
Fig.3.

(A) Fluorescence emission spectra of the wild-type Trm 5 (1.0 μM) excited at 282 nm, and the quench of the emission spectra by addition of AdoMet (100 μM) is shown. The control emission spectra of the free tryptophan solution (1.0 μM) with and without AdoMet (100 μM) were included for comparison. (B) Fitting the quench of fluorescence emission at 340 nm versus the concentration of AdoMet to a hyperbola binding equation to derive the Kd for the wild-type and P267A mutant.
To independently confirm the binding of AdoMet, a UV cross-linking assay was developed, which has been used for other AdoMet-dependent methyl transferase (31). The 5 variants that displayed lower than 20% of the normal activity were tested, together with the wild-type enzyme as a control. These enzymes were incubated at approximately the same concentration with a constant amount of 3H-labeled AdoMet, and were irradiated by long-range UV light at 312 nm. Covalent cross-linked adduct was separated from the free cofactor by SDS-PAGE analysis, and was detected by imaging the radioactivity of AdoMet. Analysis of an SDS-PAGE confirmed that the amount of each enzyme was loaded in comparable quantities (Fig.4A), while imaging of AdoMet showed that only two mutants maintained the wild-type activity of cross-links (Fig.4B). The remaining three mutants lost the cross-link activity completely (Y176A, D223A) or partially (G205A/G207A), suggesting a defective binding of AdoMet. Notably, the P267A mutant was one of the two mutants that were functional in binding the cofactor.
Fig.4.
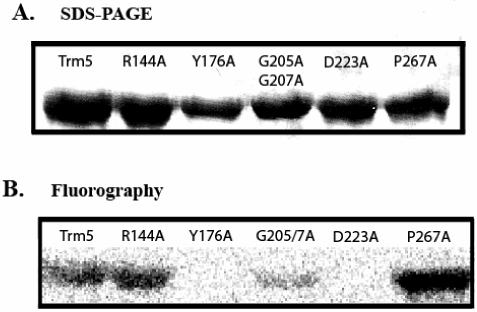
UV cross-linking of 3H-AdoMet to Trm5 and mutants examined by SDS-PAGE analysis. (A) Commassie-stained of a representative SDS-PAGE analysis of the wild-type Trm5, and the R144A, Y176A, G205A/G207A, D223A, and P267A mutants. The intensity of the stain indicates the quantity of the enzyme in the gel. (B) The SDS-PAGE used in (A) subjected to fluorography imaging of the cross-linked species.
The catalytic function of P267
The lack of strong adverse effects of the P267A mutant in binding tRNA or AdoMet suggests that the mutant was defective for catalysis. To gain further insight into the role of P267 in catalysis, we tested the possibility that the rigidity of the proline backbone may help to stabilize enzyme-substrate interactions between functional groups of N265 on the enzyme and the G37 target base in tRNA. In the crystal structure of the M·TaqI complex (30), the Asn in the NPPY motif forms hydrogen bonds to the target adenine base, and these interactions are stabilized by the first proline of the motif (NPPY). If this were the case for Trm5, we rationalized that the enzyme would be sensitive to different functional group substitutions of N265, but that this sensitivity would be abolished in the absence of the stabilizing P267.
As shown in Table 1, the N265A mutant was defective in kcat/Km for methylation by 100-fold. Two additional mutants were created that differed in functional group substitutions. The N265H mutant would maintain the same carbon skeleton as the wild-type asparagine but substitute the amide with imino nitrogen. In contrast, the second N265Q mutant would maintain the same amide nitrogen but lengthen the carbon skeleton by one methylene group. Steady-state kinetic analysis confirmed that the two mutants indeed showed marked differences in activity (Table 4). While the N265Q mutant was defective in kcat/Km by 100-fold relative to the wild-type enzyme, similar to the N265A mutant, the N265H mutant was much more active, showing a kcat/Km within 3-fold of that of the wild-type. These results demonstrate the sensitivity of Trm5 to functional group substitutions of N265, where maintenance of correct carbon skeleton is 30-fold more important than maintenance of the amide group. However, the enzyme requires minimally the imino nitrogen as a hydrogen donor because complete elimination of the donor capacity, such as in N265A, loses the 30-fold effect.
Table 4.
The kinetic relationship between N265 and P267 for tRNA methylation
| kcat (min-1) | Km(μM) | kcat/Km (μM-1min-1) | Relative kcat/Km | |
|---|---|---|---|---|
| Wild-type Trm5 | 0.5 ± 0.08 | 0.7 ± 0.03 | 0.7 ± 0.05 | 1.0 |
| N265H | 0.2 ± 0.01 | 0.8 ± 0.04 | 0.25 ± 0.02 | 0.33 |
| N265Q | 0.03 ± 0.01 | 3.2 ± 1.0 | 0.011 ± 0.006 | 0.01 |
| P267A Trm5 | 0.006 ± 0.001 | 6.0 ± 0.9 | 0.001 ± 0.001 | 0.001 |
| N265H/P267A | 0.006 ± 0 | 5.1 ± 0.1 | 0.001 ± 0.001 | 0.001 |
| N265Q/P267A | 0.007 ± 0.001 | 4.2 ± 0.4 | 0.001 ± 0.0005 | 0.001 |
The N265H and N265Q mutants were assayed by 100 and 600 nM Trm 5 with 0.5-12 and 2.5-15 μM of tRNA, respectively. The N265H/P267A and N265Q/P267A mutants were assayed by 750 nM Trm 5 with 2.5-15 tRNA. Aliquots of assay reactions were analyzed at 5, 10, 15, and 25 min.
The H and Q substitutions of N265 were then tested in the context of the P267A mutant (Table 4), where the potential stabilization by P267 was removed. In contrast to the 30-fold different sensitivity of the wild-type enzyme to these two substitutions, the P267A mutant was virtually irresponsive. Specifically, while the P267A mutant itself was defective in activity by 1000-fold compared to the wild-type enzyme (relative kcat/Km of 0.001), the double mutants N265H/P267A and N265Q/P267A showed a similar defect (relative kcat/Km of 0.002 and 0.003, respectively), demonstrating that the P267A mutant was unable to discriminate between different functional group substitutions of N265. These results are consistent with the prediction that P267 provides a stabilizing effect to functional groups of N265 during catalysis, and that when this stabilization is eliminated in the P267A mutant, the Trm5 enzyme loses the ability to discern between functional groups of N265.
Analysis of the chemical transformation rate
To directly test the role of P267 in catalysis, the wild-type enzyme and P267A mutant were compared for their catalytic ability in single turnover conditions, where the AdoMet cofactor was saturating, while enzyme was in molar excess of tRNA to allow for only one enzyme turnover. As with our studies of other tRNA enzymes (32), we termed the rate of the single turnover kinetics as the chemical transformation rate (kchem), which is a composite term that encompasses all of the rate constants from substrate binding through formation of products. For enzymes where product release might be rate-limiting, which is likely the case for Trm5, the steady-state kcat provided little information about the rate constant of the transfer step. To focus on kchem in single turnover kinetic assay, Trm5 was rapidly mixed with AdoMet and tRNA on a rapid chemical-quench instrument. Control experiments under single turnover conditions with 10 μM enzyme and limiting 1 μM tRNA showed that 50 μM AdoMet was saturating for both the wild-type and mutant enzymes. Other control experiments showed that the reaction rate was independent of the manner in which the various substrates were premixed.
To determine kchem, time courses over a range of tRNA concentrations at a fixed 10-fold molar excess of enzyme were monitored (Fig.5). For the wild-type enzyme, all time courses were well fit to a first-order exponential to determine the apparent rate constant (kapp’s) (Fig.5A). Plotting these rate constants against tRNA concentrations revealed a saturating kchem of 0.04 s-1 (Fig.5B), which is about 5-fold faster than the kcat of 0.5 min-1 (Table 1). The faster kchem than kcat supports the notion that the multiple turnover rate in steady-state kinetics is limited by product release. Importantly, analysis of the P267A mutant by single turnover kinetics clearly showed the defect of the mutant in kchem. The defect was so severe that the time courses were obtained by manual sampling of the methyl transfer reaction, rather than by the rapid quench instrument. Nonetheless, all time courses of the mutant were well fit to a first-order exponential and plotting kapp’s against tRNA concentrations showed a saturating kchem of 0.0007 s-1 (Fig.5C,D), which is nearly 60-fold slower than the kchem exhibited by the wild-type enzyme. Interestingly, the kchem of the P267A mutant is still faster than the kcat of 0.006 min-1 (0.0001 s-1) of the mutant (Table 1), suggesting that the mutation had reduced both kchem and kcat in the kinetic pathway.
Fig.5.

Determination of kchem by single turnover kinetics. (A) Time courses of the m1G37 methyl transfer reaction performed at the indicated tRNA and enzyme concentrations for the wild-type enzyme: 0.2 μM tRNA/2 μM enzyme, 0.5 μM tRNA/5 μM enzyme, 1.0 μM tRNA/10 μM enzyme, 2 μM tRNA/20 μM enzyme, 4 μM tRNA/40 μM enzyme. (B) Replot of the data by a hyperbolic fit to derive the kchem for the wild-type. (C) Time courses of the m1G37 methyl transfer reaction performed at the indicated tRNA and enzyme concentrations for the P267A enzyme: 2 μM tRNA/20 μM enzyme, 5 μM tRNA/50 μM enzyme, 10 μM tRNA/100 μM enzyme, 20 μM tRNA/200 μM enzyme, 40 μM tRNA/400 μM enzyme. (D) Replot of the data by a hyperbolic fit to derive the kchem for the P267A mutant.
Thus, comparison of single turnover kinetics of the wild-type and mutant enzymes established that the P267A mutation is defective in kchem. Because of the high enzyme and substrate concentrations used in these single turnover experiments, the second order association rates for substrates binding to enzyme are expected to be fast (33), such that the kchem measured here could indeed reflect the chemistry of the methyl transfer reaction. Alternatively, kchem might represent the rate of a slow conformational change step that follows the initial binding of substrates to enzyme. This conformational change step would necessarily involve the enzyme and both the AdoMet and tRNA substrates in a mutual induced-fit adaptation process to align reactive groups in the active site of the enzyme. In either case, it should be noted that while the P267A mutation has a significant effect on kchem, it has a minor effect on the enzyme affinity for tRNA. As shown in Fig.5B and D, the concentration of tRNA that yielded half of kchem for the wild-type enzyme is 0.7 μM, but that for the P267A mutant is 2.0 μM. This represented a 3-fold effect of the P267A mutation on tRNA binding.
Discussion
Of 18 highly conserved residues throughout the entire sequence of M. jannaschii Trm5, this study identified P267 as the most important residue for catalysis. In steady-state kinetic experiments, the P267A substitution causes the largest decrease in kcat (by 100-fold), with respect to both the tRNA- and AdoMet-dependent reactions, and additionally it causes a 10-fold defect on the Km for tRNA, such that the overall kcat/Km defect is nearly 1000-fold. In single turnover experiments that measured the rate of chemical transformation, the P267A mutation generates a severe defect on kchem, but a minor defect on tRNA binding. The relatively small effect on tRNA binding is also observed in the gel shift assay (Fig.2). Because the tRNA substrate is comparable in size to the protein enzyme, binding of tRNA likely involves the entire enzyme structure. Thus, the small effect on tRNA binding suggests that the overall global structure of the enzyme is maintained. Additioinally, because the mutant exhibits the same resistance to heat denaturation as the wild-type enzyme, it is unlikely that the mutation has perturbed enzyme stability. Collectively, these results emphasize the importance of P267 in both the chemical transformation (kchem) and catalytic turnover of Trm5 (kcat).
One important finding here is that N265, which resides in the same NLP motif as P267, makes a lesser contribution to catalysis for M. jannaschii Trm5 (Table 1). Analysis of methyl transferases that have the NPPY motif or the more relaxed form, the Asn is indispensable for some enzymatic activities but not others (34). For example, in the EcoRV adenine-N6-methyl transferase (34, 35) and the DNA:m4C methyl transferase M·NgoMXV (36), substitution of the Asn with Asp or Ser results in complete inactivation of catalysis. Similarly, the Asn in the DNA M·TaqI methyl transferase is important and substitution of this residue causes severe reduction of enzyme activity (30). Interestingly, this Asn residue stabilizes the enzyme-DNA complex by forming direct H-bonding interaction with the target base, as described by structural (29) and biophysical (37) analyses on the DNA M·TaqI methyl transferase. However, while stability is important, it is the optimal orientation of AdoMet and the target base that is most critical for catalysis. By contrast, in recent studies of the ErmC’ rRNA:m6A methyl transferase, the Asn of the similar NIPY motif is not essential for catalysis (38), which corroborates with the role of this Asn in crystal structures (39) as being involved in the stabilization of AdoMet and the target base, rather than in positioning the active site for catalysis. Another example is the yeast Tgs1 cap hypermethylase, which converts the m7G caps of snRNAs and snoRNAs to a 2,2,7-trimethyl-guanosine structure. In the analogous SPPW motif in Tgs1, alteration of the Ser residue (corresponding to Asn of the NPPY motif) to Ala has no deleterious effect, whereas alteration to Glu inactivates the enzyme activity (40). These examples demonstrate that the relative importance of the Asn residue can vary among different methyl transferases, depending on subtle variations of the catalytic pocket and on the orientation of the target base and AdoMet cofactor. In M. jannaschii Trm5, because of the lesser role of N265 in catalysis, it may perform functions more akin to those of Asn in ErmC’.
How does P267 contribute to catalysis for M. jannaschii Trm5? Prolines that reside within internal protein sequences cannot provide functional groups for a direct catalytic role, because it is a cyclic amino acid containing a secondary amine. The only known example where proline directly catalyzes the chemistry step is in the enzyme 4-oxalocrotonate tautomerase, where proline is at the N-terminus and its secondary amine serves as a general base catalyst (41). In M. jannaschii Trm5, our kinetic analysis suggests that the role of P267 must be structural, and that it exerts its role by forming an intimate structural relationship with N265 (Table 4). Specifically, while the wild-type enzyme with the intact P267 is sensitive to functional group substitutions of N265, the mutant P267A enzyme is relaxed and unable to discern among functional groups of N265. While this result emphasizes the importance of functional groups of N265, it also demonstrates that the importance is realized only in the context of P267, which supports the notion that the rigidity of P267 provides structural constraints that are important to position functional groups of N265 for correct alignment and orientation with AdoMet and the target base during catalysis. This interpretation is consistent with the unique backbone structure of proline and is in agreement with structural analysis of M·TaqI (30). It is also structurally plausible, because P267 and N265 in our homology modeling are on one side of the active-site cleft, adjacent to the G205 and G207 residues that form the binding pocket of AdoMet (Fig.1D).
The region surrounding the NLP loop that contains P267, however, is likely to have certain mobility. Indeed, structural analysis of M·TaqI indicates that although the NPPY motif itself is rigid, it is followed by a flexible loop (two residues after the NPPY motif) (42), which is unstructured in the apo form but moves into the active site when the enzyme is bound with both DNA and the cofactor (29). This movement allows the target base to flip outside of the helical center for a better accessibility to the cofactor. A similar mobility is observed in the SPOUT enzyme family. For example, E. coli TrmD has a flexible loop that contains a critical Asp residue (D169) with the potential for the acid-base catalysis of methyl transfer (16). TrmH is another member of the family and catalyzes transfer of a methyl group from AdoMet to the 2′-OH of G18 in tRNA. In a recent crystal structure of the T. thermophilus TrmH, a catalytic R41 residue is found in an unstructured loop, which has been modeled to have the ability to enter the active-site upon tRNA-binding for catalysis (43, 44). For both TrmD and TrmH, the ability of the target base to flip out into the active-site has also been suggested (43, 45). It should be noted that the active-site mobility, and rotation of the target base, are two important features that have been reported in crystal structures of DNA repair and modification enzymes (46-49). These features have also been observed in the crystal structure of tRNA pseudouridine synthase TruB and its RNA complex (50), suggesting that they provide a common catalytic strategy for enzyme modifications of nucleic acids.
The NLP motif is conserved among Trm5 proteins (Fig.1B), but is absent from TrmD proteins. Conversely, the catalytic D169 of E. coli TrmD is located in an SxxxDSF motif conserved among TrmD proteins but is absent from Trm5. Importantly, while sequences of these motifs are distinct between archaeal Trm5 and bacterial TrmD, they all contain elements of enzyme conformational flexibility.
Acknowledgement
We thank Dr. Howard Gamper for comments, and Dr. Cuiping Liu for assistance with figures.
This work was supported by a grant from the National Institutes of Health (GM66267 to YMH).
Abbreviation
- M. jannaschii
Methanocaldococcus jannaschii
- AdoMet
S-adenosyl methionine
References
- 1.Koonin EV, Mushegian AR, Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–6. [PubMed] [Google Scholar]
- 2.Forterre P. Displacement of cellular proteins by functional analogues from plasmids or viruses could explain puzzling phylogenies of many DNA informational proteins. Mol Microbiol. 1999;33:457–65. doi: 10.1046/j.1365-2958.1999.01497.x. [DOI] [PubMed] [Google Scholar]
- 3.Galperin MY, Walker DR, Koonin EV. Analogous enzymes: independent inventions in enzyme evolution. Genome Res. 1998;8:779–90. doi: 10.1101/gr.8.8.779. [DOI] [PubMed] [Google Scholar]
- 4.Ibba M, Morgan S, Curnow AW, Pridmore DR, Vothknecht UC, Gardner W, Lin W, Woese CR, Soll D. A euryarchaeal lysyl-tRNA synthetase: resemblance to class I synthetases. Science. 1997;278:1119–22. doi: 10.1126/science.278.5340.1119. [DOI] [PubMed] [Google Scholar]
- 5.Yue D, Maizels N, Weiner AM. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. Rna. 1996;2:895–908. [PMC free article] [PubMed] [Google Scholar]
- 6.Bjork GR, Kjellin-Straby K. General screening procedure for RNA modificationless mutants: isolation of Escherichia coli strains with specific defects in RNA methylation. J Bacteriol. 1978;133:499–507. doi: 10.1128/jb.133.2.499-507.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjork GR. In: Modification and editing of RNA. Benne R, editor. ASM; Washington, DC: 1998. pp. 577–581. [Google Scholar]
- 8.Bjork GR, Wikstrom PM, Bystrom AS. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science. 1989;244:986–9. doi: 10.1126/science.2471265. [DOI] [PubMed] [Google Scholar]
- 9.Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR. Improvement of reading frame maintenance is a common function for several tRNA modifications. Embo J. 2001;20:4863–73. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP. A primordial tRNA modification required for the evolution of life? Embo J. 2001;20:231–9. doi: 10.1093/emboj/20.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Dwyer K, Watts JM, Biswas S, Ambrad J, Barber M, Brule H, Petit C, Holmes DJ, Zalacain M, Holmes WM. Characterization of Streptococcus pneumoniae TrmD, a tRNA methyltransferase essential for growth. J Bacteriol. 2004;186:2346–54. doi: 10.1128/JB.186.8.2346-2354.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christian T, Evilia C, Williams S, Hou YM. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol. 2004;339:707–19. doi: 10.1016/j.jmb.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 13.Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–64. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anantharaman V, Koonin EV, Aravind L. SPOUT: a class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J Mol Microbiol Biotechnol. 2002;4:71–5. [PubMed] [Google Scholar]
- 15.Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. Embo J. 2003;22:2593–603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J Mol Biol. 2003;333:931–49. doi: 10.1016/j.jmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Wang W, Shin DH, Yokota H, Kim R, Kim SH. Crystal structure of tRNA (m1G37) methyltransferase from Aquifex aeolicus at 2.6 A resolution: a novel methyltransferase fold. Proteins. 2003;53:326–8. doi: 10.1002/prot.10479. [DOI] [PubMed] [Google Scholar]
- 18.Brule H, Elliott M, Redlak M, Zehner ZE, Holmes WM. Isolation and characterization of the human tRNA-(N1G37) methyltransferase (TRM5) and comparison to the Escherichia coli TrmD protein. Biochemistry. 2004;43:9243–55. doi: 10.1021/bi049671q. [DOI] [PubMed] [Google Scholar]
- 19.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–35. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koonin EV, Rudd KE. SpoU protein of Escherichia coli belongs to a new family of putative rRNA methylases. Nucleic Acids Res. 1993;21:5519. doi: 10.1093/nar/21.23.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783–93. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 22.King MY, Redman KL. RNA methyltransferases utilize two cysteine residues in the formation of 5-methylcytosine. Biochemistry. 2002;41:11218–25. doi: 10.1021/bi026055q. [DOI] [PubMed] [Google Scholar]
- 23.Bujnicki JM, Feder M, Ayres CL, Redman KL. Sequence-structure-function studies of tRNA:m5C methyltransferase Trm4p and its relationship to DNA:m5C and RNA:m5U methyltransferases. Nucleic Acids Res. 2004;32:2453–63. doi: 10.1093/nar/gkh564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol. 1995;253:618–32. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- 25.Huang L, Hung L, Odell M, Yokota H, Kim R, Kim SH. Structure-based experimental confirmation of biochemical function to a methyltransferase, MJ0882, from hyperthermophile Methanococcus jannaschii. J Struct Funct Genomics. 2002;2:121–7. doi: 10.1023/a:1021279113558. [DOI] [PubMed] [Google Scholar]
- 26.Tscherne JS, Nurse K, Popienick P, Ofengand J. Purification, cloning, and characterization of the 16 S RNA m2G1207 methyltransferase from Escherichia coli. J Biol Chem. 1999;274:924–9. doi: 10.1074/jbc.274.2.924. [DOI] [PubMed] [Google Scholar]
- 27.Schluckebier G, O’Gara M, Saenger W, Cheng X. Universal catalytic domain structure of AdoMet-dependent methyltransferases. J Mol Biol. 1995;247:16–20. doi: 10.1006/jmbi.1994.0117. [DOI] [PubMed] [Google Scholar]
- 28.Bujnicki JM. Phylogenomic analysis of 16S rRNA:(guanine-N2) methyltransferases suggests new family members and reveals highly conserved motifs and a domain structure similar to other nucleic acid amino-methyltransferases. Faseb J. 2000;14:2365–8. doi: 10.1096/fj.00-0076com. [DOI] [PubMed] [Google Scholar]
- 29.Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol. 2001;8:121–5. doi: 10.1038/84104. [DOI] [PubMed] [Google Scholar]
- 30.Pues H, Bleimling N, Holz B, Wolcke J, Weinhold E. Functional roles of the conserved aromatic amino acid residues at position 108 (motif IV) and position 196 (motif VIII) in base flipping and catalysis by the N6-adenine DNA methyltransferase from Thermus aquaticus. Biochemistry. 1999;38:1426–34. doi: 10.1021/bi9818016. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Cheng X. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–27. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang CM, Perona JJ, Ryu K, Francklyn C, Hou YM. Separation of two classes of tRNA synthetases by burst kinetics of aminoacylation. Nature Structural and Molecular Biology. 2006 Submitted. [Google Scholar]
- 33.Uter NT, Gruic-Sovulj I, Perona JJ. Amino acid-dependent transfer RNA affinity in a class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23966–77. doi: 10.1074/jbc.M414259200. [DOI] [PubMed] [Google Scholar]
- 34.Roth M, Helm-Kruse S, Friedrich T, Jeltsch A. Functional roles of conserved amino acid residues in DNA methyltransferases investigated by site-directed mutagenesis of the EcoRV adenine-N6-methyltransferase. J Biol Chem. 1998;273:17333–42. doi: 10.1074/jbc.273.28.17333. [DOI] [PubMed] [Google Scholar]
- 35.Roth M, Jeltsch A. Changing the target base specificity of the EcoRV DNA methyltransferase by rational de novo protein-design. Nucleic Acids Res. 2001;29:3137–44. doi: 10.1093/nar/29.15.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radlinska M, Bujnicki JM. Site-directed mutagenesis defines the catalytic aspartate in the active site of the atypical DNA: m4C methyltransferase M.NgoMXV. Acta Microbiol Pol. 2001;50:97–105. [PubMed] [Google Scholar]
- 37.Newby ZE, Lau EY, Bruice TC. A theoretical examination of the factors controlling the catalytic efficiency of the DNA-(adenine-N6)-methyltransferase from Thermus aquaticus. Proc Natl Acad Sci U S A. 2002;99:7922–7. doi: 10.1073/pnas.122231499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maravic G, Feder M, Pongor S, Flogel M, Bujnicki JM. Mutational analysis defines the roles of conserved amino acid residues in the predicted catalytic pocket of the rRNA:m6A methyltransferase ErmC’. J Mol Biol. 2003;332:99–109. doi: 10.1016/s0022-2836(03)00863-5. [DOI] [PubMed] [Google Scholar]
- 39.Schluckebier G, Zhong P, Stewart KD, Kavanaugh TJ, Abad-Zapatero C. The 2.2 A structure of the rRNA methyltransferase ErmC’ and its complexes with cofactor and cofactor analogs: implications for the reaction mechanism. J Mol Biol. 1999;289:277–91. doi: 10.1006/jmbi.1999.2788. [DOI] [PubMed] [Google Scholar]
- 40.Mouaikel J, Bujnicki JM, Tazi J, Bordonne R. Sequence-structure-function relationships of Tgs1, the yeast snRNA/snoRNA cap hypermethylase. Nucleic Acids Res. 2003;31:4899–909. doi: 10.1093/nar/gkg656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czerwinski RM, Johnson WH, Jr., Whitman CP. Kinetic and structural effects of mutations of the catalytic amino-terminal proline in 4-oxalocrotonate tautomerase. Biochemistry. 1997;36:14551–60. doi: 10.1021/bi971545h. [DOI] [PubMed] [Google Scholar]
- 42.Labahn J, Granzin J, Schluckebier G, Robinson DP, Jack WE, Schildkraut I, Saenger W. Three-dimensional structure of the adenine-specific DNA methyltransferase M.Taq I in complex with the cofactor S-adenosylmethionine. Proc Natl Acad Sci U S A. 1994;91:10957–61. doi: 10.1073/pnas.91.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure (Camb) 2004;12:593–602. doi: 10.1016/j.str.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe K, Nureki O, Fukai S, Ishii R, Okamoto H, Yokoyama S, Endo Y, Hori H. Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J Biol Chem. 2005;280:10368–77. doi: 10.1074/jbc.M411209200. [DOI] [PubMed] [Google Scholar]
- 45.Watts JM, Gabruzsk J, Holmes WM. Ligand-mediated anticodon conformational changes occur during tRNA methylation by a TrmD methyltransferase. Biochemistry. 2005;44:6629–39. doi: 10.1021/bi0481038. [DOI] [PubMed] [Google Scholar]
- 46.Vassylyev DG, Kashiwagi T, Mikami Y, Ariyoshi M, Iwai S, Ohtsuka E, Morikawa K. Atomic model of a pyrimidine dimer excision repair enzyme complexed with a DNA substrate: structural basis for damaged DNA recognition. Cell. 1995;83:773–82. doi: 10.1016/0092-8674(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 47.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–64. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 48.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–69. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 49.Reinisch KM, Chen L, Verdine GL, Lipscomb WN. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell. 1995;82:143–53. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 50.Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci U S A. 2003;100:12648–53. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]