

Abstract
The clinical efficacy of selective kinase inhibitors suggests that some cancer cells may become dependent on a single oncogene for survival. RNAi has been increasingly used to understand such “oncogene addiction” and validate new therapeutic targets. However, RNAi approaches suffer from significant off-target effects that limit their utility. Here, we combine carefully titrated lentiviral-mediated short hairpin RNA knockdown of the epidermal growth factor receptor (EGFR) with heterologous reconstitution by EGFR mutants to rigorously analyze the structural features and signaling activities that determine addiction to the mutationally activated EGFR in human lung cancer cells. EGFR dependence is differentially rescued by distinct EGFR variants and oncogenic mutants, is critically dependent on its heterodimerization partner ErbB-3, and surprisingly, does not require autophosphorylation sites in the cytoplasmic domain. Quantitative “oncogene rescue” analysis allows mechanistic dissection of oncogene addiction, and, when broadly applied, may provide functional validation for potential therapeutic targets identified through large-scale RNAi screens.
Keywords: EGF Receptor, RNAi
The concept of “oncogene addiction” has been demonstrated in a variety of mouse tumorigenesis models in which continued expression of the transforming oncogene is required for tumor maintenance (1–3). In human cancer, the clinical efficacy of imatinib confirms the essential role for BCR-ABL in Chronic Myelogenous Leukemia (4), whereas the dramatic responses of EGFR-mutant lung tumors to EGFR tyrosine kinase inhibitors (TKIs) suggest that even complex epithelial cancers harboring numerous mutations may also exhibit dependency on a single activated kinase for survival (5–7).
Although only a small number of “addicting” oncogenes have been identified to date, genome-wide approaches, including high-throughput sequencing strategies and functional RNAi screens, are underway to identify additional therapeutic targets that may be the “Achilles heels” in different types of cancer. Although large-scale nucleotide sequencing has identified candidate “driver” mutations, further functional validation is essential to establish such mutant genes as potential drug targets (8).
Gene-specific RNAi provides a powerful tool to demonstrate that specific knockdown of a mutant allele triggers cell death or proliferative arrest in oncogene-dependent cell lines. However, robust validation is required before an RNAi-mediated cell death phenotype may be viewed as proof that the targeted allele is indeed essential for cell survival (9). Confounding effects of RNAi approaches include induction of an interferon response (10), unexpected “off-target” knockdown of unrelated mRNAs (11) or saturation of endogenous microRNA pathways (12). The large number of “hits” identified in high throughput RNAi screens highlights these concerns, because many of these could represent false positives resulting from a nonspecific, toxic effect (13, 14).
To develop a robust and generalizable approach to quantify dependence on individual oncogenes, we have modeled sensitivity of lung cancer cells harboring activating mutations in EGFR to selective TKIs. By combining careful titration of EGFR knockdown vectors with the introduction of non-targetable exogenous “rescue” constructs, we directly quantified and compared the ability of various EGFR mutants to specifically substitute for endogenous EGFR in mediating cell survival. This “oncogene rescue” assay allows identification of essential downstream signaling pathways used by EGFR mutants and provides a generalizable strategy for the functional characterization and quantitative assessment of individual oncogene dependency in human cancer.
Results
Optimization of shRNA-Mediated EGFR Knockdown in Erlotinib-Sensitive Human Lung Cancer Cells.
To model EGFR dependency, we used the human non-small cell lung cancer (NSCLC)-derived PC9 cell line, harboring an amplified EGFR allele with the recurrent in-frame deletion of 15 nucleotides in exon 19 within the kinase domain (Del E746-A750 or “DEL15”) (15). As expected, PC9 cells displayed exquisite sensitivity to the EGFR TKI erlotinib, but not to a control (the MET TKI PHA-665752), thus recapitulating the known clinical phenotype of EGFR-mutant NSCLC (Fig. 1A). This was well correlated with inhibition of EGFR kinase activity, abrogation of EGFR-mediated Akt and ERK phosphorylation, and induction of apoptosis (Fig. 1 B lanes 1, 2). To compare pharmacologic EGFR kinase inhibition with EGFR knockdown, we characterized a panel of lentivirus-based shRNA constructs targeting the primary EGFR mRNA. These constructs varied in their degree of EGFR knockdown [supporting information (SI) Fig. S1 A]. The most potent shRNA vector promoted >90% cell killing (Fig. 1 A), using a concentration of viral supernatant sufficient to achieve infection of all cells (data not shown). The induction of cell death was well correlated with the extent of EGFR knockdown (Fig. S1 B). As with erlotinib, knockdown of EGFR abrogated signaling and induced apoptosis (Fig. 1 B lanes 3, 4). Thus, under these conditions, shRNA-mediated EGFR depletion phenocopies pharmacologic inhibition of EGFR kinase activity.
Fig. 1.

shRNA targeting EGFR mimicks EGFR-inhibitor treatment of cells with a kinase-activating EGFR mutation. (A) PC9 cells were treated with increasing concentrations of the EGFR TKI erlotinib or the MET inhibitor PHA-665752 (upper axis), or with increasing titers of control shRNA or shRNA targeting EGFR (lower axis). (B) Cell lysates prepared 24 h after erlotinib treatment (1 μM) or 48 h after shRNA transduction were examined by immunoblotting with the indicated antibodies.
Rescue of Oncogene Addiction by Exogenous EGFR.
Because shRNA-mediated cell death could result from specific or nonspecific effects, we established a “gene replacement strategy” to examine the ability of exogenous constructs to rescue the effects of EGFR knockdown (Fig. 2A). Wild-type and mutant EGFR cDNAs lacking the 3′-untranslated region (UTR) of the endogenous EGFR mRNA targeted by the EGFR shRNA were stably expressed in PC9 cells. Pools of cells were used to avoid clonal selection effects. As an initial test of this reconstitution assay, cells stably expressing 3′-UTR-deleted DEL15 EGFR (“DEL15-delUTR”) were transduced with a lentiviral shRNA vector targeting the 3′-UTR of the endogenous EGFR mRNA. Two days posttransduction, EGFR protein levels were virtually undetectable in control GFP-expressing cells (Fig. 2 B, lanes 1 vs. 2). However, 50% of baseline EGFR protein remained in DEL15-delUTR-reconstituted cells (lanes 3 vs. 4). Reconstituted cells also displayed significant preservation of phosphorylated Akt and ERK and suppression of PARP cleavage following shRNA treatment.
Fig. 2.

Rescue of oncogene dependence by exogenous DEL15 EGFR. (A) Schematic of rescue assay. (B) Lysates from PC9 cells stably expressing GFP or DEL15-delUTR EGFR were analyzed by immunoblot 48 h after transduction with the shEGFR vector that targets the 3′-UTR of the endogenous EGFR mRNA. (C) PC9 cells expressing GFP (blue) or DEL15-delUTR EGFR (red) were transduced with a dilution series of the shEGFR vector supernatant and then cultured in the absence (left) or presence (right) of puromycin. EGFR mutant activity was quantified by determining the amount of shRNA virus required to cause a 50% decrease in cell number (IC50) in the absence of puromycin or by calculating the AUC of virus concentration versus well intensity with puromycin. Data shown is the mean of four replicates ± 1 STDEV.
To optimize parameters for oncogene rescue, we determined the sensitivity of reconstituted PC9 cells to increasing amounts of lentiviral shRNA vector. Both GFP- and DEL15-delUTR-transduced cells were completely insensitive to the control shRNA across a broad range of concentrations (Fig. S2 A). However, the DEL15-delUTR-reconstituted cells were considerably more resistant to cell death induced by EGFR knockdown than GFP-infected cells, resulting in a 6-fold increase in the IC50 value (Fig. 2 C left). This differential sensitivity to EGFR knockdown was not due to different transduction efficiencies of the PC9 populations, as demonstrated by the acquisition of puromycin resistance following transduction with the shCTRL vector (Fig. S2 A). Hence, differential resistance to knockdown of the endogenous EGFR results from rescue by the exogenous construct.
However, the resistance to EGFR shRNA-induced cell killing was not absolute, because further increasing the amount of shRNA construct induced cell death in both GFP- and DEL15-delUTR-infected cell lines (Fig. 2 C left). Incomplete resistance to shRNA-mediated cell death could reflect incomplete reconstitution of EGFR activity, off-target toxicity at very high shRNA viral titers or prolonged expression of the shRNA (Fig. S2 B). Additionally, at low viral titers, the differential rescue by EGFR constructs was obscured by the presence of uninfected cells. Therefore, to obtain a truly quantitative measure of oncogene rescue, we used puromycin selection (i.e., the selectable marker encoded by the shRNA vector) to eliminate uninfected cells and compared surviving cell numbers at increasing shRNA viral titers across the entire range of phenotypes. By comparing the area under the curve (AUC) in a plot of cell number versus concentration of shRNA virus in the presence of puromycin, we observed a 25-fold difference between cells expressing GFP and the DEL15-delUTR EGFR variant (Fig. 2 C right). This assay proved to be highly reproducible (Fig. S3), thus providing a quantitative assessment of oncogene rescue.
Oncogene Rescue by EGFR Kinase Domain Mutants.
To identify the functional elements that mediate EGFR addiction, we first generated stable PC9 cell populations expressing a panel of EGFR variants (Table 1). These included wild-type, the kinase-inactive mutant K745A (16), and several activating mutations from patients with NSCLC, including the dual in cis T790M-DEL15 or T790M-L858R mutations, representing the acquisition of the secondary drug resistance T790M mutation within the allelic background of primary EGFR-activating mutations (17). Each variant was expressed without the endogenous 3′-UTR sequences, rendering it resistant to shRNA knockdown. The relative expression of total EGFR before and after knockdown in each cell line was similar, except for the dual in cis mutants, whose expression was consistently reduced relative to the other mutants (Fig. 3 and Fig. S4 A). As expected, EGFR remaining in cells reconstituted with the kinase-inactive variant after knockdown of endogenous protein was not phosphorylated (Fig. S4 B).
Table 1.
Summary of EGFR mutants analyzed
| Mutation | Location | Effect |
|---|---|---|
| Wild type | — | — |
| K745A | EX 19 | Inactive |
| DEL15 | EX 19 | Activating |
| DEL18 | EX 19 | Activating |
| T790M | EX 20 | Resistance |
| L858R | EX 21 | Activating |
| T790M + L858R | EX 20 + 21 | Resistance + Activating |
| T790M + DEL15 | EX 19 + 20 | Resistance + Activating |
| vIII | Ectodomain | Ligand-independent |
| DEL15 + TRUNC | EX 19 | ? |
| T790M + DEL15 + TRUNC | EX 19 + 20 | ? |
Fig. 3.

Rescue of oncogene dependence by EGFR variants found in human NSCLC. (A) EGFR levels were analyzed in PC9 cells engineered to stably express the indicated EGFR variants 2 days after transduction with the control or EGFR 3-'UTR shRNAs. (B) AUC survival analysis for PC9 cells expressing the indicated EGFR variants was determined as in Fig. 2 C. and then normalized to the activity of exogenous DEL15.
Transduction of each cell line with the control shRNA revealed similar titer-dependent survival for each reconstituted PC9 derivative in the presence of puromycin (Fig. S4 C), but striking differences in oncogene rescue (Fig. 3 B and Fig. S4 C). As expected, kinase-deficient EGFR was inactive. Wild-type EGFR exhibited weak complementing activity, as did the T790M drug resistance mutant in the context of wild-type EGFR. The NSCLC-associated activating kinase mutations displayed clear ability to rescue knockdown of endogenous DEL15 EGFR, but the L858R mutant exhibited less activity than DEL15 itself or the related DEL18 mutant. Remarkably, the combination of the two mutations DEL15 (or L858R) and T790M in cis resulted in substantially increased complementing activity compared to either mutation alone, despite lower levels of expression. Overall, rescue activity was well correlated with levels of phosphorylated Akt and ERK remaining after endogenous EGFR knockdown (Fig. S4 D). Thus, NSCLC-associated EGFR variants confer differential survival complementing activity, which is correlated with their ability to couple to downstream survival pathways. These differences correlate with the in vitro biochemical activity and transforming potential of the variant EGFR kinases (18–20).
Rescue of Mutant EGFR Dependence by Variant III EGFR.
Although wild-type EGFR lacked rescue activity, levels achieved by our expression strategy were significantly less than those present in parental PC9 cells. Therefore, we used fluorescence-activated cell sorting to isolate a cell population expressing exogenous EGFR at levels similar to the endogenous EGFR in parental PC9 cells (Fig. S5). However, higher expression of wild-type EGFR failed to protect cells from death induced by ablation of endogenous mutant EGFR (Fig. 4B), nor was it sufficient to maintain Akt or ERK phosphorylation (Fig. 4 C left). We then tested rescue activity for EGFR variant III (vIII), a constitutively activated receptor with an in-frame deletion within the ectodomain (Fig. 4 A) that endows the kinase with ligand-independent activity and is frequently detected in gliomas (21). EGFR vIII demonstrated dramatic rescue activity, comparable to that of activating kinase mutants, effectively preserving cell viability (Fig. 4 B) and downstream signaling (Fig. 4 C right) following knockdown of endogenous EGFR. Thus, a constitutively activated EGFR, but not wild-type EGFR, rescues NSCLC cells addicted to mutant EGFR kinase.
Fig. 4.

EGFR variant III rescues oncogene addiction. (A) Schematic of full-length EGFR and EGFR vIII. (B) PC9 cells stably expressing the indicated cDNAs were treated with the shEGFR vector and analyzed as in Fig. 2 C. (C) Lysates were examined by immunoblotting with the indicated antibodies after transduction of cells as in Fig. 2 B. As expected, exogenous EGFR vIII migrates with faster mobility than the endogenous full-length protein.
Cytoplasmic Phosphotyrosine Sites of EGFR are Dispensable for Oncogene Addiction.
Because rescue activity correlated with phospho-EGFR levels remaining after knockdown, we sought to determine whether the cytoplasmic phosphotyrosines of EGFR are required for oncogene addiction. We engineered an shRNA-resistant EGFR construct lacking all cytoplasmic tyrosine residues (C-terminal to Tyr 845 in the kinase activation loop), in the context of the kinase-activating mutations DEL15 and T790M-DEL15 (Fig. 5A) (22). Because deletion of these residues removes epitopes recognized by anti-EGFR antibodies, we verified similar levels of exogenous truncated and full-length receptors by flow cytometry (Fig. S6 A). Surprisingly, both EGFR-mutant truncations maintained tumor cell survival and Akt and ERK activation following depletion of endogenous EGFR, despite the absence of the C-terminal phosphotyrosines required for effector binding (Fig. 5 B, C).
Fig. 5.

Rescue of oncogene dependence does not require the EGFR cytoplasmic phosphorylation sites. (A) Schematic of EGFR showing the C-terminal tyrosine residues and the position of the truncation. (B) AUC survival analysis for PC9 cells stably expressing the indicated cDNAs was determined as in Fig. 2 C. (C) Lysates were examined by immunobloting with the indicated antibodies after transduction of cells as in Fig. 2 B. As indicated by the asterisks, removal of the C-terminal 255 aa of EGFR removes the epitope for the EGFR antibody used for immunoblot.
Because heterodimerization of EGFR with its kinase-inactive family member ErbB-3 has been proposed as a mechanism of activating downstream signaling in EGFR-addicted NSCLCs (23), we tested whether ErbB-3 activation by the truncated EGFR could explain its rescue activity. ErbB-3 potently activates phosphatidylinositol 3-kinase (PI3K) via phosphotyrosine sites in its cytoplasmic tail, which bind the PI3K p85 regulatory subunit (24). We first determined that EGFR knockdown suppressed ErbB-3 phosphorylation, and that knockdown of ErbB-3 itself suppressed downstream Akt phosphorylation (Fig. S6 B). To confirm that the C-terminal-truncated EGFR directly mediates ErbB-3-signaling to PI3K, we examined pathway activation after knockdown of endogenous EGFR in cells expressing similar levels of exogenous wild-type EGFR, full-length DEL15 EGFR or the C-terminal-truncated DEL15 variant (Fig. 6upper and Fig. S6 A). Erlotinib treatment or EGFR knockdown completely suppressed ErbB-3 and Akt phosphorylation in control (GFP) cells (lanes 1–3). EGFR knockdown also suppressed ErbB-3 and Akt activation in cells expressing exogenous wild-type EGFR (lane 6). However, phosphorylation of ErbB-3 and Akt was preserved in cells expressing full-length or C-terminally truncated exogenous DEL15 EGFR (lanes 4, 5).
Fig. 6.
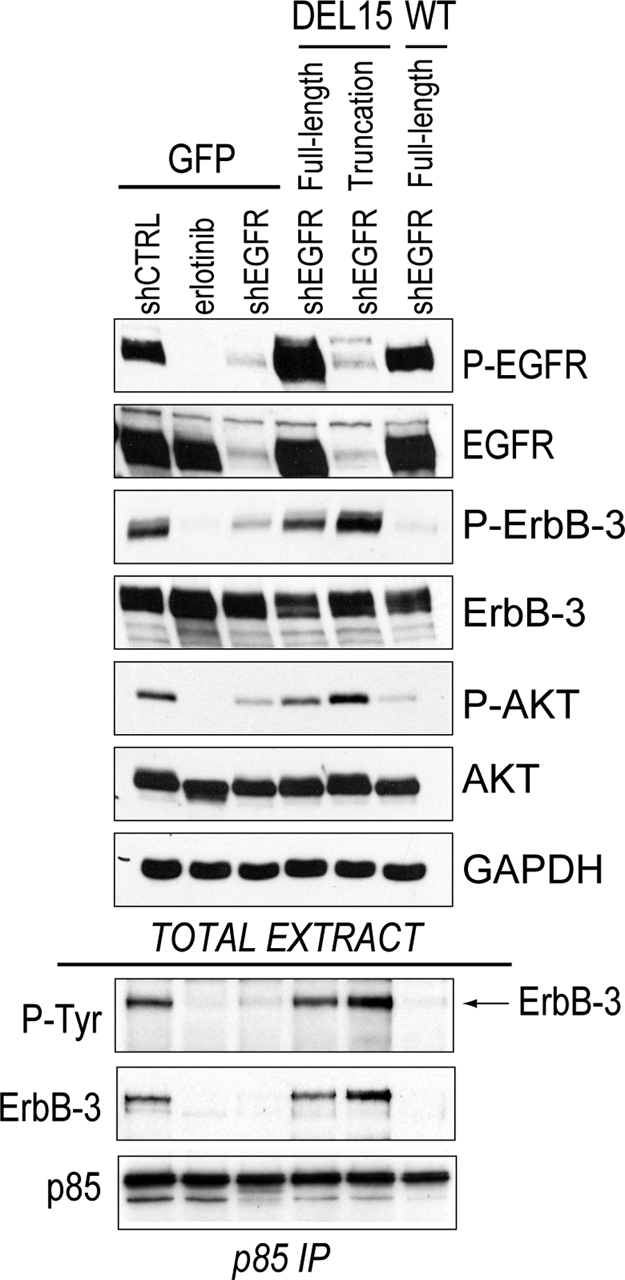
Association of PI3K with ErbB-3 does not require phosphotyrosine sites within the EGFR cytoplasmic tail. (Upper) PC9 cells expressing the indicated EGFR variants were transduced with control or EGFR shRNA vectors. At 36 h after transduction, 1 μM erlotinib was added to an aliquot of the PC9-GFP cells that had been transduced with the shCTRL vector. At 48 h after transduction, extracts were prepared and immunoblot analysis was performed with the indicated antibodies. (Lower) The p85 PI3K subunit was immunoprecipitated from 1 mg extract for each sample. Immunoprecipitates were probed with antibodies against phosphotyrosine, ErbB-3 or p85.
To confirm that DEL15 EGFR (but not wild-type) activates PI3K via ErbB-3 in the absence of EGFR's cytoplasmic tyrosines, we immunoprecipitated the p85 regulatory subunit from each cell population (Fig. 6 Lower). ErbB-3 was readily coimmunoprecipitated with p85 in control PC9 cells (lane 1), and erlotinib treatment or shRNA-depletion of the endogenous EGFR abolished this interaction (lanes 2, 3). Replacement with wild-type EGFR could not maintain the p85-ErbB-3 interaction (lane 6). However, expression of either the shRNA-resistant full-length or C-terminal-truncated DEL15 EGFR restored the ErbB-3-p85 association after endogenous EGFR loss (lanes 4, 5). Importantly, cells rescued by truncated EGFR did not demonstrate evidence of additional PI3K-associated P-Tyr proteins, suggesting that these C-terminal truncation mutants also signal primarily through ErbB-3. Thus, the cytoplasmic phosphotyrosines of EGFR are not required for EGFR addiction.
Discussion
We describe a quantitative approach for defining critical aspects of oncogene addiction in human cancer cells. Although oncogene dependence is the most appealing biological mechanism used to explain the clinical response of human tumors and the exceptional sensitivity of cell lines harboring an activated kinase to small molecule TKIs, it has largely remained a poorly understood and nonmeasurable phenomenon. This poses a particular challenge in interpreting results of large-scale kinome-wide knockdown studies, which may reveal generalized cell toxicity following suppression of many different mRNAs. Recent human cancer genome-wide sequencing studies also illustrate the difficulty in assessing the significance of mutated kinases, whose addiction potential may or may not be associated with remarkable clinical responses in future clinical trials. Our analysis indicates that carefully titrated knockdown of an endogenous kinase, combined with rescue of dependence on that kinase by its reconstituted expression, provides a quantitative assay to model oncogene addiction and to define specific signaling pathways required for such dependency.
Dissecting Dependence on Mutant EGFR Signals.
Our analysis points to reproducible differences among EGFR variants identified in gefitinib- and erlotinib-responsive EGFR mutant NSCLC. Thus, wild-type EGFR is ineffective in mediating oncogene rescue in the cells studied (when expressed at similar levels as the mutant EGFR), and of the two common activating EGFR kinase mutations, the exon 19 in-frame deletions appear to be considerably more potent than the L858R missense mutation. Of interest, a recent clinical study demonstrated that NSCLC patients with exon 19 mutations are more likely to experience durable responses to EGFR TKIs than patients with the L858R mutation (25). Our results suggest that the exon 19 mutations, which may be more catalytically active than the L858R mutation in vitro (20), are also more “addictive,” and hence their suppression by TKIs may yield greater tumor cell killing in vivo.
Although a kinase-independent role for EGFR in autophagy was recently described, the cells studied were wild-type for EGFR and did not undergo apoptosis in response to EGFR TKIs (26). In PC9 cells, any role for EGFR in preventing autophagic cell death is likely obscured by the potent apoptotic response to loss of EGFR kinase activity. Our results clearly indicate that EGFR kinase activity is critical for preventing apoptotic cell death in cells addicted to mutant EGFR.
Our results also indicate striking activity for the EGFR vIII mutant when compared to the wild-type receptor. EGFR vIII was significantly more phosphorylated than the wild-type protein, which could result in stronger activation of survival pathways and differential rescue activity. This is the first demonstration of addicting activity for EGFR vIII, because despite its frequency in glioblastomas and squamous lung cancers, patient-derived cell lines naturally expressing the endogenous mutation have never been successfully cultured. Its sensitivity to EGFR inhibitors implicates EGFR vIII as a therapeutic target in these tumors (27, 28).
Perhaps our most unexpected observation is that EGFR dependence does not require the presence of its cytoplasmic tyrosines. Instead, we found that a truncated, kinase-active EGFR lacking those residues is capable of efficient oncogene rescue by signaling primarily through ErbB-3. This pathway has been previously correlated with gefitinib sensitivity in NSCLC cell lines (23), and our analysis directly implicates signaling through ErbB-3 as a critical component of EGFR addiction. Notably, cells reconstituted with the truncated EGFR appeared more sensitive to a PI3-Kinase inhibitor than parental cells (Fig. S7), suggesting a greater dependence on this pathway for the truncated EGFR than the full-length receptor, which might partially compensate for the loss of PI3-Kinase signaling by cytoplasmic phosphotyrosine-mediated activation of alternative pathways.
Broader Implications for Genome-Wide shRNA Screens.
Despite their potential promise for uncovering new therapeutic agents, the large number of “hits” from genome-wide RNA interference screens creates a challenge for effective validation, particularly when the desired phenotype is cell death or growth arrest. Nonspecific shRNA toxicity may reflect a host of sequence, viral titer and host cell dependent effects, whereas selective oncogene addiction may itself be modulated by cellular genetic context. As such, the application of strict criteria, including rescue of dose-dependent knockdown in reconstituted cells, may be particularly important before novel targets are identified as having true therapeutic potential. As demonstrated by recent genome-wide sequencing analyses, bystander or passenger mutations commonly coexist with driver mutations in human cancers, and statistical calculations offer only an imperfect estimate of their likely functional consequence (8). Robust and reproducible oncogene reconstitution assays such as described here offer a critical measure of functional validation that should accompany the evolving annotation of the cancer genome.
Methods
DNA Constructs.
Wild-type and mutant EGFRs and GFP were cloned into pENTR223 (Invitrogen). All constructs were shuttled into lentiviral vectors 6V5 DEST (Invitrogen) or pWPI (Didier Trono, Ecole Polytechnique Federale de Lausanne, Lausanne, France) using the Gateway cloning system (Invitrogen).
shRNA Constructs and Lentiviral Production.
Lentiviral shRNA vectors were provided by William Hahn (Dana Farber Cancer Institute, Boston, MA) or were generated in our laboratory. shRNA sequences and details of lentiviral production and infection are provided in SI Methods.
Cell Lines.
See SI Methods.
Survival Assays.
Cells seeded in 96-well plates at 5–10e3 per well were transduced the following day with a dilution series of shRNA viral supernatants (0.12–12.5 μl per well). Media was exchanged 30–36 h after infection (without or with 1 μg/ml puromycin). After 5 days in culture, cells were fixed with 4% formaldehyde for 10 min and incubated with a 1:2500 dilution of Syto60 (LiCor Biosciences) for 60 min. Cell density in each well was determined with an Odyssey Infrared Imager (LiCor Biosciences), corrected for background fluorescence from empty wells and normalized to untreated wells. IC50 values for each shRNA vector were calculated by fitting the data to a sigmoid dose-response curve using nonlinear regression. Area-under-the-curve measurements were determined using the trapezoid rule.
Immunoblotting and Co-Immunoprecipitation.
Cells were plated in 6-well format at 3e5–4e5 per well. An amount of shRNA virus was used that resulted in >90% knockdown of endogenous EGFR by 48 h after transduction (typically 1 ml per well). Cell lysates were prepared in 1% Nonidet P-40 lysis buffer. For PI3K immunprecipitations (IPs), cell lysates were incubated with affinity-purified anti-p85 antibody for 4 h at 4 °C (≈5 μg per 1 mg extract) and washed 3 times with cold lysis buffer as previously described (23). Lysates and IPs were boiled in 2x Lamelli sample buffer before SDS/PAGE and transfer to PVDF membranes for immunoblot analysis. See SI Methods for details of antibodies.
Flow cytometry.
See SI Methods.
Supplementary Material
Acknowledgments.
We thank the members of the Settleman and Haber laboratories for helpful discussions. This work was supported by National Institutes of Health Grant CA115830 (JS), a V foundation award (JS), National Institutes of Health Grant CA94281 (DAH), and National Institutes of Health K08 Grant CA120060–01 (JAE). SMR is supported by a T32 Institutional Ruth L. Kirstein National Research Service Award.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. F.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0803217105/DCSupplemental.
References
- 1.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400(6743):468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 2.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 3.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Echeverri CJ, et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat Methods. 2006;3:777–779. doi: 10.1038/nmeth1006-777. [DOI] [PubMed] [Google Scholar]
- 10.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 11.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 12.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 13.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 14.Moffat J, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Bell DW, et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081–8092. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- 16.Honegger AM, et al. A mutant epidermal growth factor receptor with defective protein tyrosine kinase is unable to stimulate proto-oncogene expression and DNA synthesis. Mol Cell Biol. 1987;7:4568–4571. doi: 10.1128/mcb.7.12.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey KD, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 19.Godin-Heymann N, et al. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res. 2007;67:7319–7326. doi: 10.1158/0008-5472.CAN-06-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulloy R, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67(5):2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 21.Nishikawa R, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91(16):7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker F, et al. Activation of the Ras/mitogen-activated protein kinase pathway by kinase-defective epidermal growth factor receptors results in cell survival but not proliferation. Mol Cell Biol. 1998;18(12):7192–7204. doi: 10.1128/mcb.18.12.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelman JA, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA. 2005;102(10):3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soltoff SP, Carraway KL, III, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14(6):3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackman DM, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(13):3908–3914. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 26.Weihua Z, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13(5):385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji H, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci USA. 2006;103(20):7817–7822. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellinghoff IK, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.