

Abstract
The channel-forming component of anthrax toxin, (PA63)7, is a heptameric water-soluble protein at neutral pH, but under acidic conditions it spontaneously inserts into lipid bilayers to form a 14-stranded β-barrel ion-conducting channel. This channel plays a vital role in anthrax pathogenesis because it serves as a conduit for the membrane translocation of the two enzymatic components of anthrax toxin, lethal factor and edema factor. Anthrax channels open and close in response to changes in transmembrane voltage, a property shared by several other pore-forming toxins. We have discovered an unexpected phenomenon in cysteine-substituted channels that provides a window into this gating process: their normal voltage-dependent gating can be abolished by reaction with methanethiosulfonate (MTS) reagents or exposure to oxidizing conditions. Remarkably, this perturbation is seen with cysteines substituted at sites all along the ∼100 Å length of the channel's β-barrel. In contrast, reaction with N-ethylmaleimide, a thiol-reactive compound that does not form a mixed disulfide, does not affect gating at any of the sites tested. These findings, coupled with our biochemical detection of dimers, have led us to conclude that MTS reagents are catalyzing the formation of intersubunit disulfide bonds that lock channels in a conducting state, and that voltage gating requires a conformational change that involves the entire β-barrel.
INTRODUCTION
Cellular injury from anthrax toxin results from the action of two distinct soluble, enzymatically active subunits: lethal factor (LF) and edema factor (EF). During intoxication, protective antigen (PA; 83 kD)—anthrax toxin's pore-forming subunit—is cleaved and assembled into a heptameric “prepore” complex, and then internalized and trafficked to the late endosome with LF and EF bound to it (Young and Collier, 2007). Acidification of the endosome leads to three events that are crucial to intoxication: (1) transformation of the soluble prepore (PA63)7 into a heptameric transmembrane channel (Blaustein et al., 1989; Miller et al., 1999), (2) unfolding of EF and LF (Krantz et al., 2004), and (3) establishing a pH gradient across the endosomal membrane that helps drive the passage of LF and EF through the (PA63)7 channel (Zhang et al., 2004b; Krantz et al., 2006).
Structures of soluble conformations of PA have been determined by x-ray crystallography in both monomeric (Petosa et al., 1997) and heptameric prepore forms (Petosa et al., 1997; Lacy et al., 2004). Although the structure of the transmembrane channel form of the PA63 heptamer has not been solved, a large body of biochemical and electrophysiological data (summarized in Young and Collier [2007]) suggests that it resembles the mushroom-like structure of the heptameric pore-forming toxin α-hemolysin from Staphlococcus aureus, which has a large soluble “cap” and a 14-stranded β-barrel “stem” that forms its transmembrane domain (Song et al., 1996).
An interesting property of anthrax channels that is shared by other members of the pore-forming toxin and bacterial porin families is that they open and close in response to changes in transmembrane voltage (Donovan et al., 1981; Kagan et al., 1981; Finkelstein, 1985, 1990; Hoch et al., 1985; Blaustein et al., 1987, 1989; Gambale and Montal, 1988; Bainbridge et al., 1998). Although this process has been studied in detail in some cases, the mechanisms underlying the gating of channel-forming toxins remain largely unknown. The residues responsible for sensing the transmembrane voltage have not been identified, and the sites and extents of the actual physical gates have not been determined for these channels.
In this paper, we investigate the voltage-dependent gating of anthrax channels in planar lipid bilayers. We show that reaction of pore-lining cysteine residues in the β-barrel stem region with methanethiosulfonate (MTS) reagents leads to the formation of intersubunit disulfide bonds that lock channels in a conducting state. Because this effect occurs throughout the entire length of the β-barrel, we hypothesize that anthrax channel gating involves either a global constriction and expansion of the barrel, or a series of smaller conformational changes that propagate along the stem to a gate in the cap region. Because an open (PA63)7 channel appears to be a prerequisite for LF and EF translocation, a detailed understanding of the gating mechanism in anthrax channels may prove useful in the development of therapeutic strategies to thwart infection.
MATERIALS AND METHODS
Bacterial Strains and Plasmids
Expression plasmids encoding PA83 wild-type (WT), WT containing a C-terminal hexahistidine (His6) tag, single cysteine mutants (S312C, F313C, F314C, G317C, and G323C), and single cysteine mutants containing a C-terminal His6 tag (S290C, S329C, S330C, D335C, and E343C) were provided by A. Finkelstein (Albert Einstein College of Medicine, Bronx, NY) and R.J. Collier (Harvard Medical School, Boston, MA). The S312C-His6 construct was generated by subcloning the BamHI-XhoI fragment from pET-PAWTHis6 into identical sites in the pET-PAS312C plasmid. The pET-LFN plasmid encoding the 263–amino acid N-terminal domain of LF (LFN) containing an N-terminal His6 tag was provided by A. Finkelstein. Escherichia coli XL1-Blue cells were used in cloning, and E. coli BL21-DE3 cells were used for protein expression.
Protein Preparation
WT and single-cysteine mutants of PA (PA83) were expressed and purified as described previously (Miller et al., 1999). His6-tagged PA83 proteins were purified on a HisTrap (GE Healthcare) Ni2+-IDA column by equilibration in 20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole, pH 8, washing with 40 mM imidazole, and elution in 20 mM Tris-HCl, 500 mM NaCl, 250 mM imidazole, pH 8, followed by dialysis into 20 mM Tris-HCl, 150 mM NaCl, pH 8.5. To generate the heptameric prepore form of PA, PA83 was subjected to limited proteolysis by incubation with a 1:1,000 solution of trypsin (Sigma-Aldrich) for 30 min at room temperature, followed by quenching with 10-fold excess soybean trypsin inhibitor (Sigma-Aldrich). Nicked PA83 was loaded onto a MonoQ sepharose column (GE Healthcare) and subjected to a 10–500 mM NaCl gradient in 20 mM Tris-HCl, pH 8.5. The (PA63)7 heptameric prepore eluted at ∼400 mM NaCl and was immediately aliquoted, frozen in liquid N2, and stored at −80°C. 2 mM dithiothreitol (DTT; Sigma-Aldrich) was included in dialysis and storage buffers during preparation of the cysteine mutant proteins. The N-terminal domain of LF (LFN) was purified as described previously (Zhang et al., 2004a). The His6 tag was removed by treatment with thrombin protease (1 unit/mg His6-LFN; EMD) for 4 h at 25°C in 20 mM Tris-HCl, 150 mM NaCl, 25 mM CaCl2, pH 8.4.
Cross-linking of PA63 Channel Subunits in Liposomal Membranes
1, 2-Dioleoyl-sn-glycero-2-phosphocholine (DOPC) and 1, 2-Dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) (DOGS-NTA) were purchased from Avanti Polar Lipids, Inc. Liposomes were prepared by mixing DOPC with DOGS-NTA in chloroform at a 10:1 molar ratio. The mixture was dried under N2 gas, washed once in hexane to remove residual chloroform, and immediately resuspended in 20 mM Tris-HCl, 200 mM NaCl, pH 8.5, at a total concentration of 20 mg/ml. The lipid suspension was sonicated using a bath sonicator (Laboratory Supplies Company, Inc.) to generate small unilamellar lipid vesicles 15–50 nm in size. S312C-His6 or S330C-His6 prepore was mixed with small unilamellar lipid vesicles at a 1:20 (wt/wt) ratio and incubated for 60 min at 25°C in 20 mM Tris-HCl, 200 mM NaCl, 1 mM MgCl2, 250 μM tris(2-carboxyethyl)phosphine (TCEP), pH 8.5. After incubation, proteoliposomes were collected by centrifugation at 150,000 g for 60 min at 4°C. Pellets were washed, resuspended in TCEP-free buffer (20 mM MES, 200 mM NaCl) with a reduced pH of 6.5 to facilitate pore formation, incubated at 25°C for 60 min in the presence of hydrogen peroxide (0.3%) to facilitate disulfide formation, and maintained at 4°C overnight. Samples were then exposed to 10 mM N-ethyl maleimide (NEM) for 20 min at 25°C before denaturation to quench any free thiols, solubilized in nonreducing sample buffer in the presence of NEM (60 mM Tris-HCl, 1% SDS, 5% glycerol), and either loaded directly or boiled in 8 M urea ± 10 mM DTT at 100°C for 10 min. Protein bands were resolved by SDS-PAGE on a 4–20% Tris-HCl gradient gel (Bio-Rad Laboratories) and stained with Coomassie blue. All manipulations with S312C and S330C protein samples were performed in tandem, and resultant protein samples were run on the same gel. Gels were digitally imaged using an Image Station 2000 (Kodak).
Bilayer Formation
Planar lipid bilayers were formed using the brush technique of Mueller et al. (1963) by painting a lipid solution of 3% diphytanoyl phosphatidylcholine (DPhPC; Avanti Polar Lipids) in decane over a 250- or 500-μm aperture in a polysulfone cup. Bilayers separated two compartments (1 ml) containing buffered salt solutions (10 mM MES, 100 mM KCl, 1 mM EDTA, pH 6.6) that were stirred using small magnetic bars. Experiments were performed under voltage-clamp conditions using either a List EPC-7 or Warner BC-535 patch clamp amplifier coupled to a single pair of Ag/AgCl electrodes bathed in 3 M KCl, with electrical contact to the bath solutions made via 3-M KCl agar (3%) salt bridges. Bilayer thinning was monitored either visually or by capacitance. All voltages reported are those of the cis (protein-containing) side with respect to trans. Current responses were fed through an Axon 1322A analogue/digital converter, and recorded and analyzed on a PC using Axon pClamp software (version 8). Recordings were sampled at 10 kHz and low pass filtered at 1 kHz.
Voltage Protocols
Experiments were performed after a stable membrane conductance was obtained, typically 12–15 min after the addition of (PA63)7 to the bilayer chamber. To measure the instantaneous and “quasi” steady-state I-V relationship of WT anthrax channels, bilayers were pulsed for 2.5 s to voltages ranging from −100 to +100 mV, in 10-mV increments. Before each test pulse, the voltage was held at 0 mV to reopen any channels that may have closed during the previous pulse. Instantaneous I-V measurements were taken immediately after the start of each test pulse (after allowing for the 10–20 msec capacitance transient), and quasi steady-state I-V measurements were taken at the end of the 2.5-s test pulse.
A tail current protocol was used to obtain the apparent open probability (P0) as a function of voltage in WT and Cys mutant channels. Bilayers were subjected to a 5-s prepulse at −100 mV, followed by a 1-s test pulse, to voltages ranging from −100 to +100 mV (in 10-mV increments), and then a 1-s tail pulse to −20 mV. The apparent open probability was obtained from the normalized tail current: P0 = I/Imax, where I is the current 10 msec into the tail pulse and Imax is the maximum tail current measured. Open probability (P0) was plotted as a function of voltage and fitted to a Boltzmann equation:
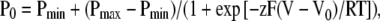 |
(1) |
where V0 is the midpoint of the voltage-activation curve, z is the effective gating charge, Pmin is the minimum open probability, V is the applied voltage, T is the temperature in degrees Kelvin, and R and F have their usual meaning.
Cysteine Modification and Oxidation Experiments
[2-(Trimethylammonium)ethyl] MTS chloride (MTSET) and N-(b-D-Glucopyranosyl)-N'-[(2-methanethiosulfonyl)ethyl] urea (MTS-glucose; Toronto Research Chemicals) were prepared as 100-mM stocks in ice-cold H2O and were aliquoted and stored at −20°C until use. NEM (Sigma-Aldrich) was prepared in H2O as a 200-mM stock and stored as described above. DTT (Sigma-Aldrich) was made fresh daily as a 200-mM stock in H2O. TCEP (Thermo Fisher Scientific) was prepared in H2O as a 250-mM stock and stored at −20°C until use. Cysteine-substituted (PA63)7 was thawed on ice and incubated with 10 mM DTT for 30 min at 25°C before addition to the bilayer chamber. In a typical recording, 1–10 fmol of protein was added to the 1-ml cis compartment of the bilayer chamber containing 250 μM TCEP to maintain cysteine thiols in reduced form during channel formation and membrane insertion. After the membrane conductance had reached a steady-state, MTSET, MTS-glucose, or NEM was added to the trans side of the bilayer while stirring the chamber. In oxidation experiments, the cis compartment was perfused with 10 volumes of DTT/TCEP-free buffer, and in certain experiments, 0.15% H2O2 (Sigma-Aldrich) was added to both cis and trans to promote oxidation.
LFN Block and Translocation Experiments
LFN (final concentration, 7.2 nM) was added to the cis side of bilayers containing either reduced or oxidized S312C channels. This led to a >95% block in conductance within 2 min under conditions of symmetric pH 6.6 and +20 mV transmembrane potential. Once conductance had reached a stable minimum, the cis compartment was perfused with 10 volumes of buffer, and the pH was adjusted to 5.6 by adding 1/30 volume 1 M MES, pH 5.2, to generate a 1-U pH gradient (cis5.6/trans6.6) while holding at +1 mV. Translocation of LFN through the channel to the trans compartment was then initiated by stepping the voltage to +20 mV, and its rate was determined by following the rise in membrane conductance.
RESULTS
Channels formed by (PA63)7 are gated by voltage; they open within 50 ms at positive voltages (Fig. 1 A) and close within seconds at voltages more negative than −20 mV (Fig. 1 C). The tendency to close at negative voltages is also evident in the region of negative slope conductance in the steady-state I-V relation compared with the instantaneous I-V, which is fairly linear (Fig. 1 D). Although not the focus of this paper, anthrax channels also exhibit a small amount of deactivation gating (e.g., closing) at larger positive voltages; this is more readily seen when pulsing from a holding potential that keeps channels open (Fig. 1 C) (Blaustein and Finkelstein, 1990; Finkelstein, 1994). To further characterize the steady-state gating properties, we applied a tail current protocol and determined the relative open probability as a function of voltage in DPhPC bilayers separating symmetric 0.1 M KCl, pH 6.6, solutions (Fig. 1, A and B). Fits of the normalized tail currents to a Boltzmann function (Eq. 1) reveal a midpoint of activation (V1/2) at −12 mV and a slope factor (z) of 1.7 elementary charges.
Figure 1.

Gating properties of WT anthrax channels in lipid bilayers. (A) Family of traces elicited by a tail current protocol using 1-s test pulses to voltages ranging from −100 to +100 mV from a holding potential of −100 mV, followed by a 1-s tail pulse to −20 mV. (B) Tail currents at −20 mV were measured 20 msec into each tail epoch to account for the capacitance transient, normalized to their maximum value, and plotted against the test pulse voltage. The points in each dataset represent the mean ± SEM of three experiments. Solid line is the fit to a Boltzmann function (Eq. 1) with V0 = −12 mV and z = 1.7. (C) Family of traces elicited by 2.5-s test pulses ranging from −100 to + 100 mV, from a holding potential of 0 mV. (D) Instantaneous current voltage measurements (circles) were taken 20 msec after each test pulse, and quasi steady-state measurements (diamonds) were taken at the end of the 2.5-s pulse. All experiments were in symmetric solutions of 20 mM MES, 100 mM KCl, 1 mM EDTA, pH 6.6.
Substitution of cysteine for pore-lining residues in the stem region does not significantly alter the gating properties of the channel under reducing conditions; a typical example is seen with S312C channels (Fig. 2 A, upper trace, and B). (We suspect that the small nonzero open probability [0.05] at large negative voltages in the unreacted S312C channel is a result of a small amount of oxidation occurring despite the presence of a reducing agent, as discussed in greater detail below.) Exposure of such a channel to the sulfhydryl-reactive reagent MTSET, however, dramatically perturbs gating—reacted channels no longer exhibit any gating at negative potentials, but instead remain essentially locked in a conducting state throughout the voltage range studied (Fig. 2 A, lower trace, and B). This can be seen both as a failure of channels to close at negative voltages, and by a lack of any turning on at positive voltages (compare upper and lower traces of Fig. 1 A). Indeed, this effect induced by MTSET unmasks the small amount of deactivation gating observed at larger positive voltages that is typically obscured by the concurrent activation gating seen in pulsing from −100 mV to the test pulse in the protocol in Fig. 1 A.
Figure 2.

Effects of MTSET on S312C channels. (A) Tail currents elicited as described in Fig. 1 A. Upper family of traces is before and lower family is 8 min after exposure to 400 μM MTSET added to the trans compartment. (B) Normalized current voltage plots were generated as described in Fig. 1 B. Unreacted S312C channels (circles) behave similarly to WT channels; solid line is Boltzmann fit (Eq. 1) with a V0 = −7 mV and z = 1.3. Tail currents measured after the addition of MTSET (squares) were normalized to their maximum current. Data points represent the mean ± SEM of three experiments. (C) The addition of 400 μM MTSET trans leads to a 75% decrease in current at +20 mV and a loss of gating at negative voltages. The breaks in the record are 70 and 170 s, respectively. (D) Current trace before exposure to MTSET depicted by dotted line; trace obtained 3 min after exposure is superimposed at the same scale (solid black line) and after normalizing to the peak current at −60 mV before reaction (gray line) to emphasize effect on gating.
The record shown in Fig. 2 C illustrates several other features of the MTSET reaction. MTSET exerts a large effect on the conductance of cysteine-substituted anthrax channels, as first shown by Benson et al. (1998). Addition of 400 μM to the trans side of the bilayer leads to an ∼75% decrease in conductance over several seconds. Reacted channels also rectify, passing more current at positive voltages than at negative voltages. This rectification, as well as the locking open effect, can be seen more clearly in Fig. 2 D, in which the current trace following MTSET exposure (solid black line) has been rescaled (solid gray line) by renormalizing it to the instantaneous current at −60 mV before MTSET addition (dotted line). Although not shown, the addition of DTT or TCEP to the chamber restores both the conductance and the voltage-gating behavior exhibited before MTSET exposure. The elimination of gating is not due to a simple shift in the voltage-activation relation to more negative potentials; as seen in the normalized I-V plot in Fig. 2 B, the relative open probability remains very high throughout the entire voltage range examined. (Although not shown, channels exhibit minimal gating even when pulsed to voltages in the −100 to −150 mV range; however, bilayers were not stable enough at these voltages for the durations necessary to construct a full normalized I-V plot.) Reaction of S312C channels with MTS-glucose, a polar, uncharged reagent, also prevents gating, although its effect on conductance is smaller and it reacts more slowly than MTSET; 400 μM MTS-glucose irreversibly reduces current by ∼40% after ∼20 s (Fig. 3).
Figure 3.

Effect of MTS-glucose on S312C channels. The addition of 400 μM MTS-glucose to the trans compartment leads to a 40% decrease in current and a loss of gating at negative voltages. The breaks in the records are 230 and 160 s, respectively.
Probing cysteines substituted at other sites along the β-barrel stem of the channel with MTS reagents reveals an interesting and altogether unexpected result: their ability to lock channels in a conducting state is not restricted to S312C or to any one particular region of the barrel. We see this same effect at every pore-lining cysteine substitution we examined throughout the entire stem region, with the sole exception of F313C, predicted to lie near the turn of the barrel (Fig. 4). Furthermore, the ability of an MTS reagent to eliminate gating is not correlated with its effects on conductance, and this is evident upon comparing the effect of a single agent at all of the sites tested, or comparing MTSET and MTS-glucose at any one site.
Figure 4.

Effects of MTSET and NEM at sites in the β-barrel transmembrane domain of the anthrax channel. Upper left is a secondary structure diagram of a β-hairpin from a single subunit, modified from Nassi et al. (2002). Residues sensitive to MTSET are indicated by solid black arrowheads, insensitive residues by unfilled arrowheads, and those not tested by open circles. Residue numbers for sites examined in this paper are shown in bold and larger type. Currents were elicited by 1-s pulses to +60 mV, followed by 10-s pulses to −60 mV. Baseline current traces before exposure to MTSET or NEM are depicted by dotted lines; traces obtained after exposure to reagent are superimposed at the same scale (solid black lines) and after normalizing to the peak currents at −60 mV before reaction (gray) to emphasize effects on gating. At all sites tested, except for F313C, MTSET exposure eliminated gating and had a significant effect on current (range, 25–98% decrease). NEM decreased conductance at many sites tested (range, 15–95% decrease) but had no effect on gating. Results are representative of three to six experiments with each reagent at each site.
NEM, another thiol-reactive reagent, also reacts with pore-lining residues throughout the stem region, although its effect on conductance is smaller than that of MTSET at each site studied (Fig. 4), with some sites showing only a minimal effect (e.g., 312, 314, 323, and 335). NEM does label these sites because washout and subsequent exposure to MTSET has little effect (not depicted). Although the overall pattern of NEM reactivity matches that of MTS reagents, its behavior is strikingly different in one important way: NEM has no effect on gating at any of the sites that we examined (Figs. 4 and 5).
Figure 5.

Effect of NEM on G317C channels. (A) The addition of 10 mM NEM to the trans compartment leads to a 95% decrease in current with no effect on gating. (B) Current trace before exposure to NEM depicted by dotted line; trace obtained 3 min after exposure is superimposed at the same scale (solid black line) and after normalizing to the peak current at −60 mV before reaction (gray line) to emphasize lack of effect on gating.
Our observation that two different MTS reagents are capable of eliminating gating, whereas NEM is not, argues that the details of the thiol chemistry play an important role in this phenomenon. Reaction of a thiol with an MTS reagent generates a mixed disulfide bond, whereas maleimides react via a Michael addition that generates a carbon–sulfur bond. This led us to investigate the notion that MTS reagents might be catalyzing thiol exchange to generate intersubunit (and intramolecular) disulfides between neighboring cysteines in the pore, as suggested previously (Zhang et al., 2004b). Consistent with this hypothesis is our observation that at certain sites in the pore, individual substitution for cysteine results in channels that are susceptible to gradual rundown in current if a reducing agent like DTT or TCEP is washed out of the chamber, an effect that can be accelerated dramatically by the addition of the oxidant H2O2 (Fig. 6 A). Furthermore, the residual current exhibits minimal gating at negative voltages (Fig. 6 B). Both the decline in current and the loss of gating could always be reversed by the readdition of reducing agent (Fig. 6). We also found that if these cysteine-substituted (PA63)7 channels are not maintained under reducing conditions, their channel activity upon addition to a bilayer bathed in DTT- or TCEP-free solutions is substantially reduced, and the addition of reducing agent leads to a large increase in current. Such observations also argue against the generation of higher oxidation states of sulfur because these are not readily reversed by these reducing agents.
Figure 6.

Effect of oxidizing conditions on S312C channels. (A) After perfusion of reducing agent from the cis compartment (first downward arrow; break in record is 250 s), there is a gradual decline in current that is dramatically accelerated by the addition of 0.15% H2O2 to both sides of the bilayer (second downward arrow). The addition of 10 mM DTT trans restores the current to the level observed before oxidation. Break in record just before DTT addition is 110 s. (B) After H2O2 exposure, channels exhibit minimal gating at −60 mV. Current traces before (solid line) and after (gray line) exposure to H2O2, with the latter normalized as described above. Trace following DTT addition is indistinguishable from baseline trace.
To test the possibility that oxidation reduces conductance by decreasing the diameter of the channel lumen, we examined its effect on the rate of protein translocation through the channel. We chose for our assay LFN, the 263-residue N-terminal fragment of LF, because the effects of voltage and pH on its rate of transport through anthrax channels have been characterized previously (Zhang et al., 2004b; Krantz et al., 2006). When LFN is added to the cis side of a bilayer containing S312C channels held at +20 mV in the presence of cis TCEP, it enters the channel and blocks the flow of the current-carrying ion (K+); this leads to the large decrease in current seen in the first portion of Fig. 7 A. If the cis pH is lowered from 6.6 to 5.6, LFN is driven completely through the channel into the trans compartment. This translocation, which can be monitored by the increase in current that reflects relief of block, takes several seconds (Fig. 7 A). In contrast, translocation of LFN is much slower through S312C channels that have been exposed to H2O2; relief of block of these channels, whose conductance is much lower, requires several minutes (Fig. 7 B). Subsequent addition of DTT to the cis compartment restores both the conductance and gating properties of the channels (Fig. 7 C), as well as the rate of LFN translocation. The effects of LFN on oxidized channels are reassuring in another regard—they provide evidence that this nongated conductance is due to properly assembled channels that possess an intact LFN docking site (Phe clamp) (Krantz et al., 2005), and not the result of some nonspecific leak induced by oxidized protein.
Figure 7.

Comparison of the rate of LFN translocation through reduced and oxidized S312C channels. (A) Reduced channels: S312C channels incorporated into the bilayer under reducing conditions (cis 250 μM TCEP) are exposed to cis LFN (7.2 nM); this leads to block of 95% of the conductance. Free LFN is then washed out of the cis compartment during the break in the record (250 s), and the pH is adjusted to 5.6 (cis5.6/trans6.6) while holding at +1 mV to prevent LFN from going through the channel. Translocation of LFN to the trans compartment is initiated by pulsing to +20 mV, and its rate is determined by following the rise in membrane conductance. (B) Same procedure as in A, except channels were oxidized before LFN addition as described in Fig. 6. The increased noise is a reflection of the marked decrease in conductance after oxidation and the increased gain. The current at 1 mV seen after the break in the record (270 s) reflects a small degree of unblocking of LFN occurring during the break. LFN translocation through oxidized channels at +20 mV proceeds much more slowly than through reduced channels. (The instantaneous current seen in pulsing from +1 mV at +20 mV is nearly ohmic, but there is rapid reblock by LFN that is not resolvable at this timescale.) Inset shows translocation progress on longer time scale. (C) The addition of 10 mM DTT to both sides of the bilayer restores both the conductance and gating properties of the channels. The recording is from the same bilayer as B and begins 185 s after the end of the inset in B. Break in the record is 155 s. Although not shown, the rate of LFN translocation is also restored after DTT exposure.
All of the experiments presented thus far in support of intersubunit disulfide formation have been electrophysiological. To further test this hypothesis, we turned to a biochemical approach that would allow us to detect disulfides by assaying for dimer formation on a gel. Miller et al. (1999) have previously shown that a reduction in the pH of a solution of purified (PA63)7 from 8.5 to 6.5 leads to an SDS-resistant form of this heptamer. To more closely mimic the state of an anthrax channel embedded in a bilayer, we reconstituted His6-tagged heptameric S312C (PA63)7 prepore complexes at high pH under reducing conditions into liposomes doped with DOGS-NTA, a lipid that contains a nickel-NTA moiety at its head group. This approach has been shown to help orient the protein at the bilayer and improve the yield of properly inserted heptameric channels after pH reduction (Sun et al., 2007). When this prepore form of the protein is exposed to denaturing conditions, it dissociates into PA63 momoners (Fig. 8, lanes 1 and 2); however, if the pH of the sample is first lowered to 6.5, a substantial portion transforms from the soluble prepore form to the heptameric SDS-resistant species (Fig. 8, lanes 3 and 6). If this transformation proceeds under oxidizing conditions (removal of TCEP, and in 0.3% H2O2) and the sample is subsequently boiled in urea, the heptameric band is no longer resolvable; however, two other bands are now present: one consistent with a monomeric species, and a higher molecular weight band consistent with dimer formation (Fig. 8, lane 4). (Although these data were generated in the presence of H2O2, similar results are obtained in its absence.) If this same sample is boiled in urea and exposed to DTT, the dimer band is no longer evident (Fig. 8, lane 5). Could the dimer band seen in lane 4 result from disulfide bond formation between exposed cysteines of denatured monomers rather than from cysteines of neighboring subunits in the SDS-resistant heptamer? There are two reasons why we think this is unlikely: (1) a high concentration of NEM is present just before and during denaturation, and this would likely quench any exposed thiols; and (2) we do not observe dimer formation in S330C mutant (PA63)7, where the cysteine is predicted to face away from the pore (Fig. 8, lanes 6–8).
Figure 8.

Intersubunit dimer formation in S312C channels. Lanes 1 and 2: At pH 8.5 under denaturing conditions, the prepore form of (PA63)7 dissociates into a single monomeric band on SDS-PAGE (lane 1, S312C; lane 2, S330C). Samples in lanes 1 and 2 were not exposed to liposomes. Lane 3: At pH 6.5, under oxidizing conditions (0.3% H2O2), heptameric S312C (PA63)7 prepore converts to the pore form and resolves as an SDS-resistant complex on SDS-PAGE. Lane 4: Upon boiling in 8 M urea, the S312C heptamer dissociates to monomeric and dimeric species. Lane 5: If the sample is boiled in the presence of 10 mM DTT, only monomeric S312C PA63 is seen. Lanes 6–8: Similar analysis of S330C, predicted to point away from the pore, yields no evidence of intersubunit dimer formation.
DISCUSSION
Over the past two decades, substantial progress has been made in understanding the role played by the PA component of anthrax toxin in the pathogenesis of Bacillus anthracis. Much of the focus has been on the assembly of PA63 into a heptameric channel, and on the function of this pore as a conduit for the membrane translocation of LF. One aspect of the behavior of anthrax channels—their opening and closing in response to changes in transmembrane voltage—has been little studied. In the course of exploring the effects of thiol-reactive compounds on pore-lining cysteine residues in these channels, we discovered an unexpected phenomenon that provides a window into this gating process: cysteine-substituted channels exposed to MTS reagents remain locked in a conducting state even at large negative voltages that would otherwise fully close unreacted channels. Three features of this gating perturbation are noteworthy: (1) The mechanism by which MTS reagents eliminate gating—catalyzing the formation of intersubunit disulfide bonds via disulfide exchange—has not been demonstrated in an ion channel, and it is different from that generally proposed for these compounds (Karlin and Akabas, 1998); (2) Reaction of cysteines at sites all along the ∼100 Å length of the channel's β-barrel generates channels that do not gate. This contrasts with results in other voltage-gated channels in which reaction at a specific site or region locks channels open (Holmgren et al., 1998; Sukhareva et al., 2003); and (3) The geometric constraints for disulfide bond formation suggest that the there must be enough flexibility along the length of the β-barrel to allow significant conformational changes to occur.
Effects of Thiol-reactive Reagents
The rationale for using an MTS reagent as a probe to study structural features of an ion channel is that reaction with a solvent-exposed cysteine introduces an adduct whose charge or bulk will affect the channel's conductance if that cysteine is coincident with or near the channel's ion conduction pathway (Karlin and Akabas, 1998). Typically, the effect of introducing a charged moiety like ethyltrimethylammonium is different than that of a neutral group like glucose, although exceptions to this have been described (Bera et al., 2002; Reeves et al., 2005). Because we find that two different MTS reagents affect gating in the same way, regardless of charge, we wondered whether the lock-open effect might arise from an action of the MTS reagent that is independent of the introduced adduct. Additional support for this notion was the observation that reaction of a single anthrax channel with either MTS-glucose or MTSET results in up to four to five successive step-wise drops in conductance (Benson et al., 1998; Zhang et al., 2004b). Because it is difficult to envision, given estimates of the size of the anthrax channel pore coupled with the size and/or charge of an -ET or -glucose adduct, that the four to five distinct chemical reactions underlying these conductance drops are the successive additions of ET or glucose groups into the channel's pore (Fig. 9 A), Zhang et al. (2004b) speculated that some of the observed step-wise drops in conductance represent disulfide exchange reactions between neighboring subunits. This mechanism is illustrated in Fig. 9 B. An initial reaction of a cysteine thiol with an MTS-X reagent generates the mixed disulfide PA-S-S-X (where PA represents one of the seven channel subunits), which then participates in thiol exchange with a cysteine on an adjacent subunit to generate an intersubunit disulfide bond between the neighboring cysteines. The addition of the S–X group into the pore generates a channel with lower unitary conductance because of steric and/or electrostatic effects. Why would generation of an intersubunit disulfide lower the conductance? Although we discuss the structural implications of disulfide formation in greater detail below, we suspect that the lumen of a channel containing an intersubunit disulfide is narrower because the cysteines on adjacent subunits that participate in a disulfide bond will be significantly closer than they would be in an open, reduced channel.
Figure 9.

(A) View through the β-barrel of α-hemolysin, with the cis end proximal. Cys was substituted for Asp at residue 127 using the “mutagenesis” feature of PyMol (with no subsequent energy minimization); this roughly corresponds to residue S312 in the PA sequence. The 11 Å distance displayed is between β carbons from adjacent subunits and represents an average of the seven nearest neighbor distances in the crystal structure of α-hemolysin. A sulfur-ethyltrimethylammonium adduct was drawn using ChemDraw, scaled appropriately, and placed in the pore at one of those residues to mimic the mixed disulfide generated by reaction of this cys residue with MTSET. (B) Proposed scheme for MTS-catalyzed intramolecular disulfide bond formation in cysteine-substituted channels. View is of a cross section of the barrel, with only a portion shown. A single MTS-X reagent reacts with one of the seven free thiols in the (PA63)7 pore to generate the mixed disulfide PA-S-S-X. This disulfide can then participate in thiol exchange with a neighboring cysteine to generate an intersubunit disulfide, releasing the S-X group in the process. Another MTS-X molecule could potentially enter the pore to react with one of the five remaining thiols to catalyze additional disulfide reactions.
There are seven potential cysteine targets in a heptameric cysteine-substituted channel, and we do not yet know how many disulfides and/or –X adducts remain in each reacted channel upon exposure to MTSET or MTS-glucose. The nature and number of reactions that take place will likely be influenced by the size and charge of the MTS reagent. We do suspect, at least for MTSET, that the final reacted state is one in which, in addition to the disulfides, an -ET group is present in the pore because such channels exhibit an outward rectification not seen in unreacted channels or channels reacted with MTS-glucose, and reaction with MTSET and MTS-glucose have different effects on conductance.
Further evidence that the effect of MTS reagents on gating is not due to the addition of an adduct in the pore is our observation that at several locations, NEM decreases conductance but never affects gating. Although MTS reagents and maleimides each react specifically with cysteine thiols, they do so using very different mechanisms. Maleimides generate a carbon–sulfur bond that cannot participate in disulfide exchange (and therefore cannot be reduced by DTT). In addition, reaction with a maleimide leaves a bulky group in the pore, yet that action does not affect gating at any of the sites we have tested.
Oxidation experiments lend additional support to our hypothesis that locking into a conducting state involves the formation of disulfide bonds. In the absence of any reducing agent, certain cysteine-substituted channels spontaneously lose their ability to gate, and this occurs much more rapidly in the presence of H2O2. Oxidized channels translocate K+ ions and proteins more slowly, demonstrating that oxidation narrows the channel's lumen. All of these effects are fully reversed by the readdition of a reducing agent, which suggests that oxidation is not decreasing conductance by generating higher oxidation states of pore-lining cysteines.
The final line of evidence in support of disulfide bond formation is our biochemical detection of dimer formation. When we analyze cysteine-substituted anthrax channels that have been reconstituted into lipid vesicles and incubated under oxidizing conditions, we are able to detect a fraction whose migration pattern on a gel is consistent with a dimer. Furthermore, this fraction is no longer resolvable if exposed to DTT, and it is not seen when reconstituting a cysteine-substituted channel whose cysteine is predicted to face away from the pore.
Structural Implications
From a structural perspective, the most compelling conclusion emerging from our findings is that the channel's entire β-barrel stem region participates in the gating process. Absent a high resolution structure of the pore form of the anthrax channel in both the closed and open states, we cannot draw a detailed picture of the gating motions from our data. Nevertheless, we can gain some insight into the magnitude of the conformational changes that might occur during gating by considering the x-ray crystal structure of α-hemolysin, which likely shares structural features with the stem domain of the anthrax channel (Song et al., 1996; Benson et al., 1998; Nassi et al., 2002; Krantz et al., 2004; Nguyen, 2004).
The luminal diameter (Cα–Cα) of α-hemolysin's β-barrel is ∼26 Å (accounting for sidechain volume, which is relevant to LF or EF translocation, yields a diameter of ∼19 Å [Krantz et al., 2004]), with residues of adjacent subunits being an average distance (Cβ–Cβ) of ∼11 Å from each other. If the β-barrel of the anthrax channel is of similar dimensions, it must be flexible enough to accommodate a sizable conformational change because disulfide bond formation requires that β carbons of participating cysteines come within 4–5 Å of each other (Careaga and Falke, 1992). What can a disulfide-containing channel that appears locked open teach us about gating motions? By trapping the channel into a nongating lower conductance state whose lumen is narrower than that of the open state, yet wider than that of the closed state, intersubunit disulfides may be capturing a gating transition on the path to the final closed configuration. Their effect would be to “straitjacket” the channel by forming a buttress that either prevents full collapse of the entire barrel, or freezes the channel into a state that doesn't allow some more subtle conformational change from propagating to a gate. Is this state a grossly distorted one that bears no resemblance to the channel's native state(s)? Several observations of reduced and oxidized channels argue against this possibility. The cation selectivity following reaction of S312C channels with MTS-glucose is essentially unchanged (reversal potential of −25 mV pre- and −24 mV post-reaction, in a fivefold cis:trans KCl gradient). This suggests that an important feature of the pore remains intact. LFN blocks reduced and oxidized channels with similar potency, which argues that oxidation does not disrupt the channel's Phe clamp, the binding site for LF located in the permeation pathway in the channel's cap domain. Finally, tetrabutylammonium blocks reduced and oxidized channels with equal potency from the cis side, lending further support to the notion that the Phe clamp is intact in oxidized channels. Thus, the only perturbation in channel structure and function that we can detect in reacted channels is a lack of appreciable gating.
K+ is the major permeant ion in our experiments, and given the size and chemical nature of the pore, it likely traverses the channel in its hydrated state (diameter of ∼8 Å). This implies that for the channel to gate shut, it must reduce its luminal diameter to less than this value somewhere along its permeation path. Although we have not yet pinpointed the exact location and extent of this gate, our preliminary examination of the state dependence of MTS-glucose reactivity suggests the presence of such a narrowing of both cis and trans to E343C (unpublished data). Because this residue appears to lie ∼3/4 of the way up the barrel (Fig. 4), and because all of the stem domain trans to this site is predicted to have β-barrel architecture, this would imply that the dynamics of gating involve a reversible change in diameter of more than a factor of 2 in some part of the barrel.
Given our observation that disulfides all along the barrel can perturb gating, and that there is gated access to residue 343 from both the cis and trans sides of the channel, we must consider the possibility that the entire barrel collapses and reexpands during gating. Although a motion of this magnitude has not been demonstrated previously in a β-barrel protein, it is worth noting that the high thermal factors in this region of the crystal structure of α-hemolysin suggest significant mobility there (Song et al., 1996). In addition, a β-barrel can change its diameter while maintaining a constant strand number if it changes its shear number (McLachlan, 1979) and perforce, the tilt of its β strands relative to the membrane normal. This phenomenon is seen in MspA, an octameric 16-stranded β-barrel porin found in the outer membrane of Mycobacterium smegmatis (Faller et al., 2004). The pore of MspA exhibits an abrupt reduction in diameter from 40 to 28 Å without changing either its β architecture or strand number; this change is associated with a drop in shear number from 32 to 16, and β-strand tilt angle from 56 to 37 degrees.
Of the MTS-sensitive residues that we have probed, only F313C shows no effect on gating upon reaction with an MTS reagent. This residue is predicted to lie at the protein–lipid interface at the very trans end of the barrel, and our failure to see a gating effect there may indicate that the trans end of the barrel is significantly wider.
Our data have allowed us to speculate about the mechanism of voltage gating in anthrax channels. A more thorough understanding of this process will ultimately require, as with any voltage-gated ion channel, that we (1) delineate the channel's gate, (2) identify the voltage-sensing residues, and (3) determine the structural connection between the sensor and the gate, goals that we are currently pursuing.
Acknowledgments
We thank Amy Sinor, Rachel Darman, Alan Finkelstein, and Dan Cox for helpful discussions, and Merritt Maduke and Chris Miller for critically reviewing the manuscript.
This work was supported by National Institutes of Health grants R01HL68985 (to R. Blaustein) and T32HL069770 (to D. Anderson).
Angus C. Nairn served as editor.
Abbreviations used in this paper: DTT, dithiothreitol; EF, edema factor; LF, lethal factor; MTS, methanethiosulfonate; MTSET, [2-(Trimethylammonium)ethyl] MTS chloride; MTS-glucose; N-(b-D-Glucopyranosyl)-N'-[(2-methanethiosulfonyl)ethyl] urea; NEM, N-ethyl maleimide; PA, protective antigen; TCEP, tris(2-carboxyethyl)phosphine; WT, wild-type.
References
- Bainbridge, G., I. Gokce, and J.H. Lakey. 1998. Voltage gating is a fundamental feature of porin and toxin beta-barrel membrane channels. FEBS Lett. 431:305–308. [DOI] [PubMed] [Google Scholar]
- Benson, E.L., P.D. Huynh, A. Finkelstein, and R.J. Collier. 1998. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 37:3941–3948. [DOI] [PubMed] [Google Scholar]
- Bera, A.K., M. Chatav, and M.H. Akabas. 2002. GABA(A) receptor M2-M3 loop secondary structure and changes in accessibility during channel gating. J. Biol. Chem. 277:43002–43010. [DOI] [PubMed] [Google Scholar]
- Blaustein, R., and A. Finkelstein. 1990. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions: effects on macroscopic conductance. J. Gen. Physiol. 96:905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein, R.O., W.J. Germann, A. Finkelstein, and B.R. DasGupta. 1987. The N-terminal half of the heavy chain of botulinum type A neurotoxin forms channels in planar phospholipid bilayers. FEBS Lett. 226:115–120. [DOI] [PubMed] [Google Scholar]
- Blaustein, R.O., T.M. Koehler, R.J. Collier, and A. Finkelstein. 1989. Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 86:2209–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga, C.L., and J.J. Falke. 1992. Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J. Mol. Biol. 226:1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan, J.J., M.I. Simon, R.K. Draper, and M. Montal. 1981. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA. 78:172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller, M., M. Niederweis, and G.E. Schulz. 2004. The structure of a mycobacterial outer-membrane channel. Science. 303:1189–1192. [DOI] [PubMed] [Google Scholar]
- Finkelstein, A. 1985. The ubiquitous presence of channels with wide lumens and their gating by voltage. Ann. NY Acad. Sci. 456:26–32. [DOI] [PubMed] [Google Scholar]
- Finkelstein, A. 1990. Channels formed in phospholipid bilayer membranes by diphtheria, tetanus, botulinum and anthrax toxin. J. Physiol. (Paris). 84:188–190. [PubMed] [Google Scholar]
- Finkelstein, A. 1994. The channel formed in planar lipid bilayers by the protective antigen component of anthrax toxin. Toxicology. 87:29–41. [DOI] [PubMed] [Google Scholar]
- Gambale, F., and M. Montal. 1988. Characterization of the channel properties of tetanus toxin in planar lipid bilayers. Biophys. J. 53:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch, D.H., M. Romero-Mira, B.E. Ehrlich, A. Finkelstein, B.R. DasGupta, and L.L. Simpson. 1985. Channels formed by botulinum, tetanus, and diphtheria toxins in planar lipid bilayers: relevance to translocation of proteins across membranes. Proc. Natl. Acad. Sci. USA. 82:1692–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, M., K.S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit disulfide bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- Kagan, B.L., A. Finkelstein, and M. Colombini. 1981. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc. Natl. Acad. Sci. USA. 78:4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, A., and M.H. Akabas. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145. [DOI] [PubMed] [Google Scholar]
- Krantz, B.A., A.D. Trivedi, K. Cunningham, K.A. Christensen, and R.J. Collier. 2004. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J. Mol. Biol. 344:739–756. [DOI] [PubMed] [Google Scholar]
- Krantz, B.A., R.A. Melnyk, S. Zhang, S.J. Juris, D.B. Lacy, Z. Wu, A. Finkelstein, and R.J. Collier. 2005. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 309:777–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz, B.A., A. Finkelstein, and R.J. Collier. 2006. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 355:968–979. [DOI] [PubMed] [Google Scholar]
- Lacy, D.B., D.J. Wigelsworth, R.A. Melnyk, S.C. Harrison, and R.J. Collier. 2004. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA. 101:13147–13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan, A.D. 1979. Gene duplications in the structural evolution of chymotrypsin. J. Mol. Biol. 128:49–79. [DOI] [PubMed] [Google Scholar]
- Miller, C.J., J.L. Elliott, and R.J. Collier. 1999. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 38:10432–10441. [DOI] [PubMed] [Google Scholar]
- Mueller, P., D.O. Rudin, H.T. Tien, and W.C. Westcott. 1963. Methods for the formation of single bimolecular lipid membranes in aqueous solution. J. Phys. Chem. 67:534–535. [Google Scholar]
- Nassi, S., R.J. Collier, and A. Finkelstein. 2002. PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry. 41:1445–1450. [DOI] [PubMed] [Google Scholar]
- Nguyen, T.L. 2004. Three-dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. J. Biomol. Struct. Dyn. 22:253–265. [DOI] [PubMed] [Google Scholar]
- Petosa, C., R.J. Collier, K.R. Klimpel, S.H. Leppla, and R.C. Liddington. 1997. Crystal structure of the anthrax toxin protective antigen. Nature. 385:833–838. [DOI] [PubMed] [Google Scholar]
- Reeves, D.C., M. Jansen, M. Bali, T. Lemster, and M.H. Akabas. 2005. A role for the beta 1-beta 2 loop in the gating of 5-HT3 receptors. J. Neurosci. 25:9358–9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L., M.R. Hobaugh, C. Shustak, S. Cheley, H. Bayley, and J.E. Gouaux. 1996. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 274:1859–1866. [DOI] [PubMed] [Google Scholar]
- Sukhareva, M., D.H. Hackos, and K.J. Swartz. 2003. Constitutive activation of the Shaker Kv channel. J. Gen. Physiol. 122:541–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J., G. Vernier, D.J. Wigelsworth, and R.J. Collier. 2007. Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J. Biol. Chem. 282:1059–1065. [DOI] [PubMed] [Google Scholar]
- Young, J.A., and R.J. Collier. 2007. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 76:243–265. [DOI] [PubMed] [Google Scholar]
- Zhang, S., A. Finkelstein, and R.J. Collier. 2004. a. Evidence that translocation of anthrax toxin's lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc. Natl. Acad. Sci. USA. 101:16756–16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., E. Udho, Z. Wu, R.J. Collier, and A. Finkelstein. 2004. b. Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys. J. 87:3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]