

Abstract
Gossypol, a cottonseed extract derivative, acts as a BH3-mimetic, binding to the BH3 pocket of antiapoptotic proteins and displacing pro-death partners to induce apoptosis. However, knowledge on the molecular underpinnings of its downstream effects is limited. Since chronic lymphocytic leukemia (CLL) cells express high levels of antiapoptotic proteins that act as a survival mechanism for these replicationally quiescent lymphocytes, we investigated whether gossypol induces apoptosis in these cells and what mechanism underlies gossypol-mediated cytotoxicity. Gossypol induced cell death in a concentration- and time-dependent manner; 24-hour incubation with 30 μM gossypol resulted in 50% cell death (median; range, 10%-80%; n = 47) that was not abrogated by pan-specific caspase inhibitor. Starting at 4 hours, the mitochondrial outer membrane was significantly permeabilized (median, 77%; range, 54%-93%; n = 15). Mitochondrial outer membrane permeabiliztaion (MOMP) was concurrent with increased production of reactive oxygen species (ROS); however, antioxidants did not abrogate gossypol-induced cell death. Mitochondrial membrane permeabilization was also associated with loss of intracellular adenosine triphosphate (ATP), activation of BAX, and release of cytochrome c and apoptosis-inducing factor (AIF), which was translocated to the nucleus. Blocking AIF translocation resulted in a decreased apoptosis, suggesting that AIF contributes to gossypol-mediated cytotoxicity in CLL lymphocytes.
Introduction
Gossypol, a natural product derived from cottonseed extracts, was originally extensively investigated in China as a male contraceptive agent.1 It exhibits a type of enantiomerism that arises from restricted rotation: the (−)- gossypol isomer showed greater cytotoxicity than the (+)- isomer in several human cancer cell lines.2 To reduce toxicity, structural modifications in the (−)- gossypol isomer led to the analog apogossypol, which lacks the reactive aldehydic groups and displays proapoptotic activity comparable with that of gossypol3; another derivative, gossypolone, demonstrates lower cytotoxicity than the parent compound.4 The success of these agents resulted in the development of additional analogs, such as the l-isomer of gossypol, AT-101,5 and ABT-737, which was designed on the basis of structure activity-based studies.6,7
The gossypol isomers and analogs exhibited inhibitory activity against a wide range of human carcinoma cell lines and in tumor xenograft models.8–11 However, these and other investigations illustrated different actions of this agent, and knowledge on the molecular underpinnings of its biologic effects is still limited.
Early studies in HeLa cells demonstrated that the cytotoxicity of gossypol was based on its inhibitory activity on DNA synthesis through DNA polymerases alpha and beta12 or on topoisomerases.13 Gossypol's effect on DNA synthesis, leading to S-phase arrest, was specific: no effects on RNA and protein syntheses were observed.14 In concordance with the concept of DNA-directed actions, gossypol appears to play a significant role in the killing of tumor cells by inhibiting cellular proliferation15,16 and by modulating the expression of cell cycle–regulatory proteins Rb and cyclin D1 in human mammary cancer cells and in cyclin D1–transfected human fibrosarcoma cells.17 Consistently, (−)-gossypol caused growth suppression of human prostate cancer cells by down-regulation of cyclin D1, Rb, CDK4, and CDK6 and by up-regulation of p21 and TGF-beta1 at the mRNA and/or protein levels.18,19 Taken together, these findings elucidate the cell cycle– and DNA-directed actions of this agent.
The recent discovery that gossypol mimics a BH3 binding protein has focused investigations on the role of this alkaloid on antiapoptotic proteins. For example, nuclear magnetic resonance–based methods and fluorescence polarization displacement assays have established that gossypol and its analogs bind and interact with the antiapoptotic proteins BCL-2 and BCL-XL.20 In concert with this observation, gossypol induced apoptosis in colon carcinoma cells ectopically expressing BCL-2 and BCL-XL.21 Similarly, gossypol treatment induced cytochrome c release and BAK activation in whole cells as well as in isolated mitochondria that expressed high levels of BCL-2.22 In PC-3 cells, gossypol induced apoptosis by inhibiting the heterodimerization of BCL-XL/BCL-2 with proapoptotic molecules which involves the release of apoptosis-inducing factor (AIF) from mitochondria into the cytosol.23 Further studies showed that gossypol could also induce BAX/BAK-independent activation of apoptosis and cytochrome c release via a conformational change in BCL-2.24 Recent work on head and neck squamous cell carcinoma cells with high endogenous BCL-XL demonstrated that gossypol binds to this protein and that the apoptosis was related to the ratio of BCL-XL and BCL-XS.25 In HL-60 cells, the cytotoxicity occurs through mitochondrial dysfunction pathway.26 In addition to this mitochondrial celldeath pathway, an extrinsic cell death pathway throughup-regulation of Fas/Fas ligand has been reported.27 Taken together, these recent findings identified gossypol as a BH3 mimetic with profound cytotoxicity in cells that express a high level of anti-apoptotic proteins.
These investigations were done in rapidly dividing cell lines, making it challenging to interpret gossypol's exclusive effect on antiapoptotic proteins since DNA-dependent and cell cycle–regulated actions may also play a role in cell death.
Circulating mature B lymphocytes from patients with chronic lymphocytic leukemia (CLL) are replicationally quiescent, accumulate in bone marrow and blood due to their long survival,28 and represent an example of a malignancy caused by failure of programmed cell death.29 Therefore, we hypothesized that these cells are an ideal model system for investigating the cytotoxicity and mechanism of action of gossypol.30
Methods
A racemic mixture of gossypol, mainly the (−)- isoform, was provided by Bioenvision, New York, NY. Z-VAD.fmk was obtained from MP Biomedicals (Solon, OH), glutathione was purchased from Sigma-Aldrich (St Louis, MO), calpeptin was obtained from EMD Biosciences (La Jolla, CA), and N-acetyl cysteine was procured from Calbiochem (San Diego, CA). All other chemicals were reagent grade.
Patients
Leukemic lymphocytes obtained from patients with CLL (n = 50) were incubated in vitro with gossypol and used for different pharmacological, biochemical, and molecular end points. Most patients (70%) were previously untreated and generally had a high white blood cell count in the peripheral blood (Table S1 available on the Blood website; see the Supplemental Materials link at the top of the online article). All patients provided written informed consent to participate in this laboratory protocol, which was approved by the institutional review board of the University of Texas M. D. Anderson Cancer Center in accordance with the Declaration of Helsinki.
Isolation of lymphocytes
Whole blood was collected in heparinized tubes and processed to obtain mononuclear cells (leukemic lymphocytes). Cells were washed twice with cold phosphate-buffered saline (PBS) and resuspended in 10 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum. A Coulter Channelyzer (Coulter Electronics, Hialeah, FL) was used to determine the cell number and mean cell volume. The lymphocytes were resuspended at a concentration of 1 × 107 cells/mL and were used fresh for all experiments.
Incubation of lymphocytes
Lymphocytes were incubated with gossypol at the indicated concentrations and times for different assays. To inhibit caspases, the pan-specific caspase inhibitor Z-VAD.fmk was used at 50 μM, and to inhibit the generation of ROS, N-acetyl cysteine (NAC; 1 mM) was used. Cells were incubated with these inhibitors 2 hours before the addition of gossypol, and this was followed by 24-hour incubation with both the inhibitors and gossypol.
Apoptosis assays
Apoptosis was measured in annexin V binding assay using a detection kit I from Pharmingen (San Diego, CA) according to the manufacturer's instructions. Briefly, cells were washed with PBS and resuspended in 200 μL of 1 × annexin binding buffer (BD Biosciences, Franklin Lakes, NJ) at a concentration of 1 × 106 cells/mL. Annexin V–fluorescein isothiocyanate (FITC; 5 μL) was added, and the cells were incubated in the dark for 15 minutes at room temperature. To these labeled cells, 10 μL propidium iodide (50 μg/mL) was added, and flow cytometry was performed immediately (FACSCalibur; Becton, Dickinson, San Jose, CA). Data from at least 10 000 events per sample were recorded and processed using CellQuest software (Becton Dickinson). As another measure of apoptosis, poly(ADP-ribose) polymerase (PARP) cleavage was measured by immunoblotting.
Quantitation of cellular ATP pool
Before and after gossypol treatment, the cells were processed to extract nucleotides. The cellular adenosine triphosphate (ATP) pool was determined using a high-pressure liquid chromatography procedure as described before.31 The cellular ATP concentration was between 3 and 4 mM in untreated CLL lymphocytes. Data were expressed as the percentage of the control concentration after drug treatment.
Determination of mitochondrial outer membrane permeabilization
Before and after gossypol treatment, 106 cells were washed in PBS, resuspended in medium, and incubated with tetramethylrhodamine methyl ester (TMRM; Invitrogen, Carlsbad, CA) and FITC-conjugated annexin V in the dark for 15 minutes at room temperature.32 Samples were analyzed using a FACSCalibur flow cytometer. (FL1 = annexin V–FITC; FL2 = TMRM.) Data from at least 10 000 events per sample were recorded and processed using CellQuest software (Becton Dickinson).
Measurement of superoxide generation
The superoxide-meditated oxidation-sensitive fluorogenic dye dihydroethidium (DHE; Invitrogen Molecular Probes, Eugene, OR) was used to evaluate intracellular production of superoxide radicals. DHE is cell- permeable and, in the presence of O2, it is oxidized to fluorescent ethidium, which intercalates into DNA. Briefly, cells were washed once with serum- and phenol red–free medium at the indicated time point and then incubated with 5 μM DHE for 30 minutes in medium. The fluorescence of ethidium was measured using a FACscan flow cytometer supplied with CellQuest software.
Nuclear, cytosolic, and mitochondrial protein extraction
Cytosolic and mitochondrial fractions were isolated from treated and untreated cells. Briefly, 2 × 107 cells were harvested, washed once with cold PBS, and resuspended in 3 volumes of isolation buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 70 mM sucrose, 210 mM mannitol, and protease inhibitors). After incubating in an ice bath for 10 minutes, the cell suspension was homogenized with 100 strokes in a 2-mL glass homogenizer using pestle A (Dounce homogenizer; Bellco Glass, Vineland, NJ). The samples were centrifuged at 1500g at 4°C for 5 minutes to remove nuclei pellet. The supernatants were centrifuged at 15 000g for 15 minutes to separate the mitochondrial (pellet) and cytosolic fractions (supernatant). Mitochondrial pellets were lysed with TNC buffer33 (10 mM Tris acetate, pH 8.0, 0.5% NP-40, 5 mM CaCl2).
Total cell extraction
Cells were lysed on ice for 20 minutes in lysis buffer containing 25 mM HEPES, pH 7.5, 300 mM NaCl, 1.5 mM MgCl2, 0.5% sodium deoxycholate, 20 mM glycerophosphate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.2 mM EDTA, pH 8, 0.5 mM dithiothreitol, 1 mM sodium orthovanadate, pH 10, and protease inhibitor. Cells were centrifuged at 14 000g for 10 minutes at 4°C, and the supernatant was removed and stored at –80°C until use.
Immunoblot analysis
Protein content was determined using a DC protein assay kit (Bio-Rad Laboratories, Hercules, CA). Aliquots (30 μg) of total cell protein were boiled with Laemmli sample buffer and loaded onto 8% to 12% sodium dodecyl sulfate–polyacrylamide gels and transferred to nitrocellulose membranes (GE Osmonics Labstore, Minnetonka, MN). Membranes were blocked for 1 hour in PBS-Tween-20 containing 5% nonfat dried milk and then incubated with primary antibodies for 2 hours, followed by species-specific horseradish peroxidase–conjugated secondary antibody (diluted 1:5000) for 1 hour. The blots were visualized by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL) and normalized to the actin (Sigma-Aldrich), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam, Cambridge, MA), Anti-porin (VDAC; Calbiochem, San Diego, CA), and Histone H1 mouse monoclonal (sc-8030) levels in each extract. Rabbit polyclonal antibody to MCL-1 (sc-819), mouse monoclonal antibody to BCL-2 (sc-509), rabbit polyclonal antibody to BCL-XL, mouse monoclonal antibody to cytochrome c, rabbit polyclonal antibody to AIF (BD Pharmingen International, San Diego, CA; amino acids 517-531 and Santa Cruz Biotechnology Laboratories, Santa Cruz, CA; amino acids 1-300), mouse monoclonal antibody to Bax (obtained from Santa Cruz Biotechnology Laboratories, Santa Cruz, CA), and mouse monoclonal antibody to PARP (from BD Pharmingen International, San Diego, CA) were used to detect these proteins in each sample.
Immunohistochemistry and confocal microscopy
Untreated and treated CLL cells were centrifugally applied to slides using cytospin and fixed in 100% methanol. The slides were washed with PBS and resuspended in PBS-Tween-20 and incubated with anti-AIF antibody at a 1:50 dilution in 3% goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA) and incubated for 1 hour at 37°C. Next, the cells were washed in PBS and incubated for 1 hour with DAPI (4,6 diamidino-2-phenylindole) and fluorescent-conjugated secondary antibody (1:100 dilution), followed by washing with PBS and resuspending in PBS-Tween-20. The cell nuclei were stained with DAPI and slides were covered with glass cover slips and sealed with clear nail polish. The cells were examined for fluorescence using a Fluoview FV500 confocal microscope system (Olympus, Center Valley, PA) with a 60× objective. The projections of single slices were saved as TIF files.
Results
Gossypol induced apoptosis in CLL lymphocytes
Gossypol binds with high affinity to the BH3 binding pocket of antiapoptotic BCL-2 family members.3,20 Since Bcl-2 family proteins are highly expressed in CLL lymphocytes, we determined whether these cells were susceptible to gossypol treatment (Figure 1Ai,ii). When untreated CLL lymphocytes were incubated for 24 hours, 22% apoptotic cells (spontaneous apoptosis); increased to 63% after gossypol treatment. Similar studies were done in samples from 47 patients, and data from 9 patients are presented (Figure 1Aiii). In all cases, cell death was induced in a median of 50% of the cells (range, 10%-80%; n = 47). Apoptotic effect of gossypol was not associated with Rai stage of the disease, prior treatment, ZAP-70 or immunoglobulin status, or IgVH mutation. Similarly 5 patients who had a refractory disease also responded to gossypol treatment. In addition, p53 status, chromosomal trisomy, and ATM mutation, although rarely observed among these 50 patients, did not change extent of apoptosis (Table S1).
Figure 1.

Gossypol-mediated CLL lymphocyte cell death and effect of caspase inhibitor or an antioxidant. (A) Gossypol induces cell death in chronic lymphocytic leukemia (CLL) lymphocytes. CLL primary cells were cultured for 24 hours either (i) alone (control) or (ii) with 30 μM gossypol, and the cell viability was determined by flow cytometry after double staining with FITC-conjugated annexin V and propidium iodide. (iii) Percentage of cell death in CLL primary cells from 9 individual patients treated with gossypol for the indicated times. (iv) CLL primary cells were incubated with 10 and 30 μM gossypol for the indicated times. Cells were lysed and cleavage of PARP was measured by immunoblotting. Actin was used as a loading control. Similar experiments were done in 8 patients, and data from 2 representative patients are shown. (B) Effect of caspase inhibitor on gossypol-induced cell death. CLL primary cells were incubated (i) for 24 hours alone (control), (ii) with Z-VAD.fmk, (iii) with 30 μM gossypol plus Z-VAD.fmk, or (iv) with 30 μM gossypol for 24 hours, and then the cell viability was determined as described in panel A. (v) Percentage of cell death in CLL primary cells from 9 individual patients treated with Z-VAD.fmk and gossypol (C, untreated control; Z, Z-VAD.fmk; Z + G, Z-VAD.fmk plus gossypol; G, gossypol). (C) Effect of antioxidant (NAC) on gossypol (G)–induced cell death. CLL primary cells were incubated (i) for 24 hours alone (control), (ii) with NAC, (iii) with 30 μM gossypol plus NAC, or (iv) with 30 μM gossypol, and then the cell viability was determined as described in panel A. (v) Percentage of cell death in CLL primary cells from 6 individual patients treated with NAC plus gossypol (Con, untreated control).
As another measure of apoptosis, PARP cleavage was analyzed by immunoreaction. As shown for 2 representative samples (Figure 1Aiv), PARP cleavage was apparent at 4 or 8 hours after treatment with 30 or 10 μM gossypol, respectively.
Gossypol-induced cell death was caspase-independent
To determine whether gossypol-induced apoptosis was dependent on caspases, CLL cells (n = 13) were incubated for 24 hours both alone and with gossypol in both the presence and absence of a pan-caspase inhibitor, Z-VAD.fmk. Similar to untreated cells, cells treated with Z-VAD.fmk alone did not undergo any significant cell death (7% and 9%, respectively). However, incubation of CLL cells with Z-VAD.fmk did not abrogate gossypol-induced cell death (70% and 88%, respectively) in these lymphocytes (Figure 1Bi-iv). When the same concentration of Z-VAD.fmk was tested with fludarabine, as a positive control (10 μM) there was inhibition of apoptosis (without Z-VAD.fmk 15%-25%; with Z-VAD.fmk 7%-15%; n = 3; data not shown) in CLL cells. Similar studies were done in samples from 13 patients, and representative data from 9 patients are presented in Figure 1Bv. Overall, Z-VAD.fmk had no effect in 10 of 13 samples and provided only minor protection in the other 3 samples, suggesting that in most cases, gossypol-induced cell death was caspase-independent.
To assess the role of caspases during early apoptosis, leukemic lymphocytes from 3 patients were incubated with and without Z-VAD.fmk in both the presence and absence of gossypol for 4, 8, and 24 hours. Even at these early time points, Z-VAD.fmk did not abrogate gossypol-induced cell death (Table S2). To further establish that gossypol-induced apoptosis is caspase-independent, we incubated CLL cells for 24 hours with 10 μM and 30 μM gossypol in both the presence and absence of Z-VAD.fmk and monitored them for cleavage of PARP protein (Figure S1). Once again, the presence of Z-VAD.fmk did not protect PARP from gossypol-induced cleavage.
Gossypol-mediated cell death was ROS-independent
Execution of apoptosis by phenolic compounds such as gossypol is closely linked to the disruption of mitochondrial electron transport coupled with the production of superoxide radicals. Thus we tested whether an antioxidant like NAC or glutathione can abrogate gossypol-induced cell death. CLL primary cells (n = 6) were incubated with and without NAC in both the presence and absence of gossypol. NAC alone did not induce significant cell death relative to that in untreated control cells (18% and 21%, respectively). Preincubation of CLL cells with NAC did not abrogate gossypol-induced cell death (75% and 82%, respectively; Figure 1Ci-iv). Data were similar in additional samples (Figure 1Cv) and were also consistent when the other antioxidant, glutathione (1 mM; 2-hour pretreatment), was used (data not shown).
Gossypol induced MOMP in CLL lymphocytes
To determine whether gossypol binding to BCL-2 family proteins would affect mitochondrial outer membrane permeabilization (MOMP), lymphocytes were incubated either alone or with gossypol, and MOMP was measured using TMRM. Untreated cells that tested positive for TMRM become negative for TMRM as the membrane became permeable, as can be seen in Figure 2Ai. Similar studies were done in 15 patient samples, and data from 5 representative patients are presented (Figure 2Aii). In all patients, there was a significant MOMP, with a median of 77% TMRM negativity (range, 54%-93%; n = 15).
Figure 2.

MOMP and effect of caspase inhibitor or an antioxidant on MOMP in gossypol-treated CLL lymphocytes. (A) Gossypol induces mitochondrial outer membrane permeabiliztaionn (MOMP) in CLL lymphocytes. (i) Cells were incubated for 24 hours either alone (control) or with 30 μM gossypol, and the MOMP was measured by flow cytometry using a double-staining (TMRM and FITC-conjugated annexin V). (ii) Percentage of MOMP in CLL primary cells from 5 patient samples treated with gossypol. (B) Effect of a caspase inhibitor on gossypol-induced MOMP. CLL cells were incubated for 24 hours alone (C, control), with Z-VAD.fmk (Z), with 30 μM gossypol (G) plus Z, or with gossypol (G), and the MOMP was measured by flow cytometry as described in panel A. Percentages of MOMP in CLL primary cells from 6 patients are provided. (C) Effect of antioxidant (NAC) on gossypol-induced MOMP. CLL cells were incubated for 24 hours alone (C, control), with NAC, with gossypol (G) plus NAC, or with 30 μM gossypol, and then the MOMP was measured as described in panel A. Percentages of MOMP in CLL primary cells from 6 individual patients are provided.
Gossypol-induced MOMP was caspase- and ROS-independent
Next we assessed whether Z-VAD.fmk could abrogate the MOMP caused by gossypol in CLL primary cells (n = 15). As in untreated cells, cells treated with Z-VAD.fmk alone did not undergo significant permeabilization. Treatment with gossypol alone and with Z-VAD + gossypol resulted in a similar extent of MOMP, suggesting that the gossypol-induced MOMP is caspase independent (Figure 2B). Similarly, preincubation of CLL cells with NAC (1 mM) did not protect the cells from gossypol-induced MOMP, as shown for 6 patients' samples (Figure 2C). These results were similar in all patients, suggesting that superoxide production is also not the mechanism underlying the gossypol-induced MOMP. Furthermore, preincubation with the other antioxidant, glutathione, did not have any protective effect on gossypol-induced MOMP in these lymphocytes (data not shown). These data suggest that the mitochondrial membrane is one of the direct targets of gossypol.
Temporal relationship between cell death and MOMP
To elucidate whether the MOMP and induction of cell death occur in parallel or consecutively, we compared time-dependent cell death (Figure 3A) and MOMP (Figure 3B). Data from the same 3 patients' samples suggested that cell death remained nearly at basal levels after 4 and 8 hours but increased dramatically at 24 hours. In contrast, MOMP was quantifiable as early as 4 hours and gradually increased with time, suggesting that MOMP is an earlier event, whereas cell death follows later. To further ascertain the MOMP, the intracellular concentration of endogenous ATP and its loss were quantitated before and after treatment with gossypol for various times (Figure S2). The ATP pool decreased from 3 to 4 mM (100% value) to less than 100 μM during the 24 hours of incubation in a time-dependent manner. The loss was apparent as early as 4 hours. Again, this dissipation of ATP was not due to cell death; instead, it was due to MOMP, which occurred in as early as 4 hours.
Figure 3.

Temporal relationship between apoptosis and MOMP in gossypol-treated CLL lymphocytes. Time-dependent cell death (A) and MOMP (B) were compared in CLL cells from 3 different patients after incubation with 30 μM gossypol at the indicated times. Cell death and MOMP were measured as described in Figures 1 and 2, respectively.
Gossypol increased the production of superoxide in CLL lymphocytes
Given the paradoxic effects of phenolic compounds in oxidative stress,34 previous studies have evaluated the pro-oxidant property of gossypol in the production of superoxide radicals.35–37 When we incubated lymphocytes for 24 hours either alone or with 30 μM gossypol, the production of superoxide radicals increased significantly (Figure 4A). Furthermore, as in the untreated cells (Figure 4Bi), superoxide production was not increased significantly by either Z-VAD.fmk (Figure 4Bii) or NAC (Figure 4Biii), and neither of those agents abrogated gossypol-induced superoxide production (Figure 4Biv-vi). Results were similar in samples from 15 patients.
Figure 4.

Gossypol-mediated superoxide production and effect of caspase inhibitor or antioxidant on superoxide production in CLL lymphocytes. (A) Gossypol increases the production of superoxide in CLL lymphocytes. CLL primary cells were incubated for 24 hours either (i) alone or (ii) with 30 μM gossypol for 24 hours, and the superoxide production was measured by flow cytometry using hydroethydine (HEt). (B) Effect of a caspase inhibitor and an antioxidant on gossypol-induced superoxide production. CLL primary cells were incubated for 24 hours (i) alone, (ii) with Z-VAD.fmk, (iii) with NAC, (iv) with 30 μM gossypol plus Z-VAD.fmk, (v) with 30 μM gossypol plus NAC, or (vi) with 30 μM gossypol, and then the superoxide production was measured.
Effect of gossypol on the levels of antiapoptotic proteins
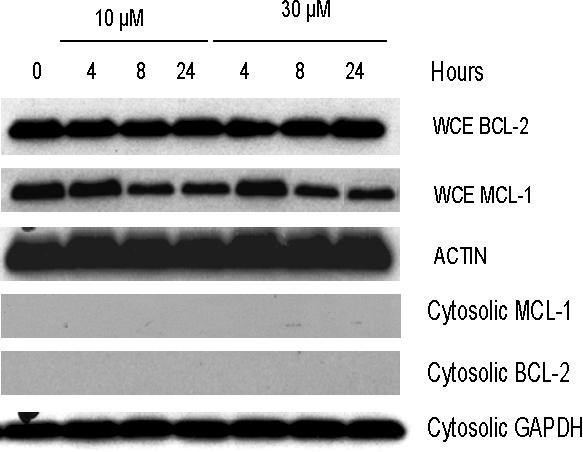
To determine whether gossypol modulates the expression level of proteins important in the survival of B-CLL cells, we focused on the expression of the antiapoptotic proteins MCL-1, BCL-2, and BCL-XL. Gossypol did not affect the expression of these proteins at early time points (4 and 8 hours) but caused a decrease (ranging from minor to profound) in the level of MCL-1 at 24 hours. Similar experiments were done in samples from 8 patients, and data from 3 representative patients are provided (Figure 5A-C). Since gossypol is a BH3 mimetic, we wanted to test if gossypol treatment and its interactions with antiapoptotic proteins cause displacement of MCL-1 and BCL-2 into cytosol. Immunoblot data (Figure S3) confirmed that these antiapoptotic proteins were not displaced into cytosol at any time after gossypol treatment, although there was abundant of these proteins present in whole cell extract (WCE; Figure S3).
Figure 5.

Effect of gossypol on the antiapoptotic proteins of Bcl-2 family members. CLL primary cells were incubated with 10 μM and 30 μM gossypol for the indicated times. Cells were lysed and immunoblotted using antibodies that recognize Mcl-1, Bcl-2, and Bcl-xl. Actin was used as a loading control. Similar experiments were done in 8 patients; data from 3 representative patients are provided (A-C). In panel C the last lane is spliced from the same gel that was originally loaded on lane 8.
BAX translocation after gossypol treatment
BAX, a proapoptotic protein, has been shown to be the key factor for activation of the intrinsic cell death pathway, which occurs after the binding of BH3 small-molecule inhibitors to members of the BCL-2 family. To test whether that also happens with gossypol, CLL cells were treated with 2 concentrations of gossypol, and the mitochondrial and cytosolic fractions were separated. As Figure 6A illustrates, the level of BAX in the cytosol decreased after treatment and it is translocated to mitochondria in concentration and time dependent manner after treatment with gossypol, suggesting that BAX activation is involved in the release of proapoptotic molecules.
Figure 6.

Effect of gossypol on release and translocation of proapoptotic proteins. (A) Gossypol induces the release of proapoptotic proteins in CLL primary cells. CLL primary cells were treated with 10 μM or 30 μM gossypol for indicated hours, and the expression of cytochrome c (CYTO C), BAX (in mitochondrial BAX, the last 3 lanes were spliced from the same gel which was originally loaded on the first 3 lanes. A vertical line is inserted to indicate a repositioned gel lane, and apoptosis-inducing factor (AIF) ∼57-kDa (amino acids-1-300) and AIF ∼67-kDa (amino acids 517-531) were analyzed. GAPDH, VDAC, and Histone 1 (cytosolic, mitochondrial, and nuclear loading controls, respectively) were evaluated by immunoblotting in cytosolic (C) and mitochondrial (M) fractions. Similar experiments were done in 5 patients, and the immunoblot from 1 representative patient is provided. (B) Translocation of AIF to the nucleus in gossypol-treated CLL primary cells. CLL primary cells were treated with 10 μM or 30 μM gossypol for 4, 8, and 24 hours, and the expression of AIF (the last lane is spliced from the same gel which was originally loaded on lane 8) was analyzed in the nuclear fraction by immunoblotting. GAPDH, VDAC, and histone 1 (cytosolic, mitochondrial, and nuclear loading controls, respectively) were used as loading controls. (C) CLL primary cells were incubated with 10 μM and 30 μM gossypol for 24 hours, stained with anti-AIF antibody, and then subjected to fluorescent staining with DAPI (nuclear staining, blue) and FITC-conjugated secondary antibody (green), and the images were captured on confocal microscopy.
Effect of gossypol on release of proapoptotic protein
Because gossypol treatment resulted in the MOMP, we examined the fate of proapoptotic proteins that are localized in the inter-membranous space of the mitochondria. When CLL lymphocytes were incubated with 10 μM and 30 μM gossypol, the cytochrome c level decreased in the mitochondrial fraction but simultaneously increased in the cytosol (Figure 6A first 2 rows). The release of cytochrome c was both time and dose dependent. Similar experiments were done in samples from 3 patients.
Loss of AIF from mitochondria and translocation to nucleus
Immunoblotting showed that initially, the amount of AIF in the mitochondrial fraction of untreated cells was abundant, but it decreased significantly after treatment with gossypol for 24 hours (Figure 6A). It has been shown that AIF is synthesized as an ∼ 67-kDa protein with an N-terminal presequence of 101 residues that is proteolytically removed upon translocation into the mitochondria to generate the mature ∼ 57-kDa protein.38 In our studies we tested for both of these AIF fragments using different antibodies and interestingly AIF ∼ 67-kDa was not detected in the cytosol; however, there was release of AIF ∼ 57-kDa into cytosol in concentration and time dependent manner after gossypol treatment. This finding is consistent with other reports.38,39 To ascertain the fate of AIF, we performed immunoblotting on the nuclear fraction of untreated and gossypol-treated cells. The results showed that the mitochondrial AIF ∼ 67-kDa translocated into the nucleus gradually by 24 hours after treatment with 30 μM gossypol (Figure 6B).
Immunohistochemical staining further established that AIF translocates from the mitochondria to the nucleus. CLL cells were incubated with 10 μM and 30 μM gossypol for 24 hours, stained with DAPI and anti-AIF antibody, and then examined with a confocal microscope. In gossypol-treated cells, AIF translocated into the nucleus in a concentration-dependent manner. This testing was performed in samples from 4 patients; data from 1 representative patient is presented in Figure 6C.
Effect of calpeptin, a calpain inhibitor, on gossypol-induced AIF release
The release of AIF into the cytosol is associated with caspase-independent apoptosis and occurs after calpain activation.40 Since our studies showed that gossypol-induced apoptosis was caspase independent, we focused on the vital role of AIF in gossypol-induced apoptosis. To test this, we incubated CLL primary cells for 24 hours alone, with calpeptin, with calpeptin for 2 hours followed by gossypol, or with gossypol alone and then examined AIF ∼ 67-kDa expression in the cytosolic, mitochondrial, and nuclear fractions (Figure 7). In the presence of calpeptin, significantly less AIF was released into the cytosol than with gossypol alone, suggesting that gossypol-induced cell death is at least partly mediated by AIF. To further confirm this effect, cell death studies were done after treatment with gossypol in presence and absence of calpeptin. However, there was only partial inhibition of apoptosis induced by gossypol in presence of calpeptin (data not shown) suggesting that either AIF mediated pathway is responsible for partial cell death or when this pathway is blocked additional factors contribute to gossypol-induced cell death.
Figure 7.

Calpeptin, a calpain inhibitor, partially inhibits AIF release in gossypol-treated CLL primary cells. CLL primary cells were either untreated (U) or treated with 30 μM gossypol (G) in the presence and absence of calpeptin (Cp) for 24 hours, and the expression of AIF was evaluated by immunoblotting in the cytosolic (C), mitochondrial (M), and nuclear (N) fractions. Histone 1 (H1) was used as loading control.
Discussion
In this study, we demonstrated that gossypol induced apoptosis in CLL primary cells. Because primary CLL lymphocytes are quiescent, gossypol's DNA synthesis and cell cycle–directed actions would not occur in this replicationally null background system. Since gossypol is an antiapoptotic BCL-2 family antagonist, and the pattern of BCL-2 family gene expression in leukemic CD5+ B-CLL cells is skewed toward prevention of apoptosis and the relentless accumulation of CD5+ leukemic B cells, this disease type provides an ideal system for investigating the actions of gossypol that stem from inhibition of BCL-2 antiapoptotic family members. With this respect, the apoptosis41,42 was observed in all samples studied, albeit at different levels, which was not dependent on the disease (Rai) stage, prior therapy, refractory disease, chromosomal abnormalities, ZAP70, and IgVH mutation status.
Phenolic compounds such as gossypol induce apoptosis by the production of ROS.34 ROS and mitochondria play an important role in apoptosis induction under both physiologic and pathologic conditions. Oxidation of the mitochondrial pores by ROS may contribute to cytochrome c release caused by disruption of the mitochondrial membrane potential.43 Although gossypol increased the production of ROS in CLL lymphocytes in our study, it was not the central mediator of apoptosis because antioxidants were unable to circumvent the gossypol-mediated cell death. Hence, ROS generation appears to be an effect rather than the cause of gossypol-induced cell death.
Although gossypol-induced apoptosis appeared to involve the intrinsic cell death pathway,41,42 which seems to originate in the mitochondria, the induction of apoptosis was not caused by a decrease in the expression level of BCL-2 pro-survival family members. Multiple death stimuli, with or without the involvement of the classical BCL-2 family, converge on the mitochondria to trigger the death cascade.44–46 When CLL lymphocytes were treated with gossypol, MOMP was seen in every patient sample tested. It is possible that gossypol binds to prosurvival BCL-2 family members,20 thereby making these proteins unavailable to protect the integrity of the mitochondrial outer membrane. In fact, several pieces of evidence suggested that the permeability of the mitochondrial outer membrane is the primary apoptosis-triggering event. First, MOMP was an early event, starting as early as 4 hours after treatment with gossypol. Second, this occurred much earlier than cell death did, and it was not inhibited by Z-VAD.fmk (Figure 3A,B). Third, MOMP was not affected by NAC and or glutathione. Finally, permeability of the mitochondrial membrane resulted in the release of proapoptotic molecules from the mitochondria, which could initiate the cell death cascade.47,48 It is established that BCL-2 family proteins regulate the MOMP and thereby control the release of apoptosis-inducing proteins that are sequestered in the inter-membrane space of mitochondria.49,50 Three proteins that are specifically associated with mitochondria and cell death are BAX, cytochrome c, and AIF.
The BAX protein resides in the cytosol of resting cells in an inactive state and, in response to various stimuli, it undergoes specific conformational changes that allow its targeting and insertion into the mitochondrial outer membrane, where it forms a pore that allows the release of proapoptotic factors into the cytosol.51,52 This permeabilization function of BAX is dependent on the BAX-associated production of ROS.53 Consistent with these findings, our data showed that when CLL lymphocytes were incubated with 30 μM gossypol, BAX translocated from the cytosol to the mitochondria and proapoptotic proteins such as cytochrome c and AIF were liberated in as early as 4 hours (Figure 6A).
Release of cytochrome c from the mitochondria is regulated by BCL-2 family proteins with the essential requirement of BAX/BAK.54–56 Cytochrome c release, which we also observed in this study, has been identified as the key event in the formation of apoptosomes and the activation of caspase 9.57 Although we had insufficient numbers of cells to investigate caspase activation, it is highly likely that at least in part; gossypol-induced cell death involves the apoptosome-mediated death cascade. Because inhibition of caspases did not affect cell death generated by gossypol, the apoptosomal pathway does not appear to be the primary mechanism of lymphocyte demise.
The most notable factor involved in the induction of apoptosis, AIF, which is a death-executing molecule that induces caspase-independent cell death,58 is released into the cytosol through mitochondrial permeability transition pores that involve voltage-dependent anion channels in the outer membrane.59–63 Under pathologic conditions, AIF is released from the mitochondria and translocated into the nucleus, where it induces large-scale DNA fragmentation.58 Translocation of AIF into the nucleus in dying cells has been observed in many models of disease and cell death.64–66 It has been shown that AIF is synthesized as an ∼ 67-kDa protein with an N-terminal presequence of 101 residues that is proteolytically removed upon translocation into the mitochondria to generate the mature ∼ 57-kDa protein.38,39 In concert with those findings, in our studies when we tested for both of these AIF fragments, AIF ∼ 67-kDa was not detected in the cytosol; however, there was release of AIF ∼ 57-kDa into cytosol in concentration and time dependent manner after gossypol treatment. Because AIF acts as an endonuclease, it does not require an active caspase system. Since caspase inhibition did not protect the cells against gossypol-induced death, it is highly likely that AIF is the primary executor of cell death in CLL lymphocytes after gossypol treatment. As in previous studies, which reported that inhibition of calpain activation by calpeptin precluded AIF release, we also found that the presence of calpeptin and CaCl2 inhibited the release of AIF, demonstrating that proteolytic activity was required for its release.40
In conclusion, our data in CLL lymphocytes obtained from 50 patients with CLL demonstrated that gossypol is an effective cytotoxic agent for this disease. The chief and initiating event appears to be MOMP, which leads to the release of proapoptotic proteins. Primary among these proteins is AIF, which may be responsible for the caspase-independent cell death of these replicationally quiescent CLL lymphocytes.
Supplementary Material
Acknowledgments
The authors thank Karen F. Phillips for critically reading and reviewing the manuscript.
This work was supported in part by grants from the National Cancer Institute, Department of Health and Human Services (CA57629, CA81534, and Cancer Center Support Grant P30-16672), CLL Global Research Foundation–US/European Alliance, and Bioenvision, Inc.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.B. designed and performed all of the experiments, analyzed data, and wrote the manuscript; W.G.W. identified patients with CLL who were used in the study and is the principal investigator of the laboratory protocol to obtain blood samples from patients; M.J.K. identified patients with CLL who were used in the study; and V.G. conceptualized the research, directed K.B. in the design of experiments and analyses of data, and finalized the text of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Varsha Gandhi, Department of Experimental Therapeutics, Unit 71, The University of Texas M. D. Anderson Cancer Center, Houston, TX 77030; e-mail: vgandhi@mdanderson.org.
References
- 1.Wu D. An overview of the clinical pharmacology and therapeutic potential of gossypol as a male contraceptive agent and in gynaecological disease. Drugs. 1989;38:333–341. doi: 10.2165/00003495-198938030-00001. [DOI] [PubMed] [Google Scholar]
- 2.Dodou K, Anderson RJ, Small DA, Groundwater PW. Investigations on gossypol: past and present developments. Expert Opin Investig Drugs. 2005;14:1419–1434. doi: 10.1517/13543784.14.11.1419. [DOI] [PubMed] [Google Scholar]
- 3.Becattini B, Kitada S, Leone M, et al. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem Biol. 2004;11:389–395. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 4.Dodou K, Anderson RJ, Lough WJ, Small DA, Shelley MD, Groundwater PW. Synthesis of gossypol atropisomers and derivatives and evaluation of their anti-proliferative and anti-oxidant activity. Bioorg Med Chem. 2005;13:4228–4237. doi: 10.1016/j.bmc.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 5.Xu L, Yang D, Wang S, et al. (−)-Gossypol enhances response to radiation therapy and results in tumor regression of human prostate cancer. Mol Cancer Ther. 2005;4:197–205. [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu YW, Chik CL, Knazek RA. An in vitro and in vivo study of antitumor effects of gossypol on human SW-13 adrenocortical carcinoma. Cancer Res. 1989;49:3754–3758. [PubMed] [Google Scholar]
- 9.Stein RC, Joseph AE, Matlin SA, Cunningham DC, Ford HT, Coombes RC. A preliminary clinical study of gossypol in advanced human cancer. Cancer Chemother Pharmacol. 1992;30:480–482. doi: 10.1007/BF00685601. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert NE, O'Reilly JE, Chang CJ, Lin YC, Brueggemeier RW. Antiproliferative activity of gossypol and gossypolone on human breast cancer cells. Life Sci. 1995;57:61–67. doi: 10.1016/0024-3205(95)00243-y. [DOI] [PubMed] [Google Scholar]
- 11.Le Blanc M, Russo J, Kudelka AP, Smith JA. An in vitro study of inhibitory activity of gossypol, a cottonseed extract, in human carcinoma cell lines. Pharmacol Res. 2002;46:551–555. doi: 10.1016/s104366180200230x. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg LJ, Adlakha RC, Desai DM, Rao PN. Inhibition of DNA polymerase alpha by gossypol. Biochim Biophys Acta. 1986;866:258–267. doi: 10.1016/0167-4781(86)90051-5. [DOI] [PubMed] [Google Scholar]
- 13.Adlakha RC, Ashorn CL, Chan D, Zwelling LA. Modulation of 4′-(9-acridinylamino)methanesulfon-m-anisidide-induced, topoisomerase II-mediated DNA cleavage by gossypol. Cancer Res. 1989;49:2052–2058. [PubMed] [Google Scholar]
- 14.Wang Y, Rao PN. Effect of gossypol on DNA synthesis and cell cycle progression of mammalian cells in vitro. Cancer Res. 1984;44:35–38. [PubMed] [Google Scholar]
- 15.Kable EP, Parsons PG. Potency, selectivity and cell cycle dependence of catechols in human tumour cells in vitro. Biochem Pharmacol. 1988;37:1711–1715. doi: 10.1016/0006-2952(88)90433-9. [DOI] [PubMed] [Google Scholar]
- 16.Thomas M, von Hagen V, Moustafa Y, Montmasson MP, Monet JD. Effects of gossypol on the cell cycle phases in T-47D human breast cancer cells. Anticancer Res. 1991;11:1469–1475. [PubMed] [Google Scholar]
- 17.Ligueros M, Jeoung D, Tang B, Hochhauser D, Reidenberg MM, Sonenberg M. Gossypol inhibition of mitosis, cyclin D1 and Rb protein in human mammary cancer cells and cyclin-D1 transfected human fibrosarcoma cells. Br J Cancer. 1997;76:21–28. doi: 10.1038/bjc.1997.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang YW, Wang LS, Chang HL, et al. Effects of serum on (-)-gossypol-suppressed growth in human prostate cancer cells. Anticancer Res. 2006;26:3613–3620. [PubMed] [Google Scholar]
- 19.Van Poznak C, Seidman AD, Reidenberg MM, et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat. 2001;66:239–248. doi: 10.1023/a:1010686204736. [DOI] [PubMed] [Google Scholar]
- 20.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 21.Zhang M, Liu H, Guo R, et al. Molecular mechanism of gossypol-induced cell growth inhibition and cell death of HT-29 human colon carcinoma cells. Biochem Pharmacol. 2003;66:93–103. doi: 10.1016/s0006-2952(03)00248-x. [DOI] [PubMed] [Google Scholar]
- 22.Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE. (−)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- 23.Zhang M, Liu H, Tian Z, Griffith BN, Ji M, Li QQ. Gossypol induces apoptosis in human PC-3 prostate cancer cells by modulating caspase-dependent and caspase-independent cell death pathways. Life Sci. 2007;80:767–774. doi: 10.1016/j.lfs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Lei X, Chen Y, Du G, et al. Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J. 2006;20:2147–2149. doi: 10.1096/fj.05-5665fje. [DOI] [PubMed] [Google Scholar]
- 25.Oliver CL, Bauer JA, Wolter KG, et al. In vitro effects of the BH3 mimetic, (-)-gossypol, on head and neck squamous cell carcinoma cells. Clin Cancer Res. 2004;10:7757–7763. doi: 10.1158/1078-0432.CCR-04-0551. [DOI] [PubMed] [Google Scholar]
- 26.Hou DX, Uto T, Tong X, et al. Involvement of reactive oxygen species-independent mitochondrial pathway in gossypol-induced apoptosis. Arch Biochem Biophys. 2004;428:179–187. doi: 10.1016/j.abb.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Chang JS, Hsu YL, Kuo PL, Chiang LC, Lin CC. Upregulation of Fas/Fas ligand-mediated apoptosis by gossypol in an immortalized human alveolar lung cancer cell line. Clin Exp Pharmacol Physiol. 2004;31:716–722. doi: 10.1111/j.1440-1681.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 28.Bentley DP, Pepper CJ. The apoptotic pathway: a target for therapy in chronic lymphocytic leukemia. Hematol Oncol. 2000;18:87–98. doi: 10.1002/1099-1069(200009)18:3<87::aid-hon661>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 29.Kaufmann SH, Gores GJ. Apoptosis in cancer: cause and cure. Bioessays. 2000;22:1007–1017. doi: 10.1002/1521-1878(200011)22:11<1007::AID-BIES7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Gottardi D, Alfarano A, De Leo AM, et al. In leukaemic CD5+ B cells the expression of BCL-2 gene family is shifted toward protection from apoptosis. Br J Haematol. 1996;94:612–618. doi: 10.1046/j.1365-2141.1996.d01-1856.x. [DOI] [PubMed] [Google Scholar]
- 31.Balakrishnan K, Stellrecht CM, Genini D, et al. Cell death of bioenergetically compromised and transcriptionally challenged CLL lymphocytes by chlorinated ATP. Blood. 2005;105:4455–4462. doi: 10.1182/blood-2004-05-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rasola A, Geuna M. A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry. 2001;45:151–157. doi: 10.1002/1097-0320(20011001)45:2<151::aid-cyto1157>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 33.Achanta G, Sasaki R, Feng L, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li AS, Bandy B, Tsang SS, Davison AJ. DNA-breaking versus DNA-protecting activity of four phenolic compounds in vitro. Free Radic Res. 2000;33:551–566. doi: 10.1080/10715760000301091. [DOI] [PubMed] [Google Scholar]
- 35.Benhaim P, Mathes SJ, Hunt TK, Scheuenstuhl H, Benz CC. Induction of neutrophil Mac-1 integrin expression and superoxide production by the medicinal plant extract gossypol. Inflammation. 1994;18:443–458. doi: 10.1007/BF01560692. [DOI] [PubMed] [Google Scholar]
- 36.Wu DF, Yu YW. Superoxide free radical formation stimulated by (+/−), (+), (−) gossypol in rat liver and kidney microsomes. Proc Chin Acad Med Sci Peking Union Med Coll. 1986;1:150–156. [PubMed] [Google Scholar]
- 37.de Peyster A, Quintanilha A, Packer L, Smith MT. Oxygen radical formation induced by gossypol in rat liver microsomes and human sperm. Biochem Biophys Res Commun. 1984;118:573–579. doi: 10.1016/0006-291x(84)91341-x. [DOI] [PubMed] [Google Scholar]
- 38.Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 39.Loeffler M, Daugas E, Susin SA, et al. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 40.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 41.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann SH, Hengartner MO. Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001;11:526–534. doi: 10.1016/s0962-8924(01)02173-0. [DOI] [PubMed] [Google Scholar]
- 43.Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 45.Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene. 2002;21:8786–8803. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- 47.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 48.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 49.Gottlieb RA. Mitochondria: execution central. FEBS Lett. 2000;482:6–12. doi: 10.1016/s0014-5793(00)02010-x. [DOI] [PubMed] [Google Scholar]
- 50.Daniel PT, Schulze-Osthoff K, Belka C, Guner D. Guardians of cell death: the Bcl-2 family proteins. Essays Biochem. 2003;39:73–88. doi: 10.1042/bse0390073. [DOI] [PubMed] [Google Scholar]
- 51.Polster BM, Kinnally KW, Fiskum G. BH3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J Biol Chem. 2001;276:37887–37894. doi: 10.1074/jbc.M104552200. [DOI] [PubMed] [Google Scholar]
- 52.Hetz C, Vitte PA, Bombrun A, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 53.Kirkland RA, Franklin JL. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid Redox Signal. 2003;5:589–596. doi: 10.1089/152308603770310257. [DOI] [PubMed] [Google Scholar]
- 54.Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 55.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antonsson B, Conti F, Ciavatta A, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 57.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 58.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 59.Scarlett JL, Murphy MP. Release of apoptogenic proteins from the mitochondrial intermembrane space during the mitochondrial permeability transition. FEBS Lett. 1997;418:282–286. doi: 10.1016/s0014-5793(97)01391-4. [DOI] [PubMed] [Google Scholar]
- 60.Andreyev A, Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- 61.Susin SA, Lorenzo HK, Zamzami N, et al. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 63.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 64.Cao G, Clark RS, Pei W, et al. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- 65.Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X, Chen J, Graham SH, et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}