

Abstract
The modulatory role of whole cardiac myosin binding protein-C (cMyBP-C) on myosin force and motion generation was assessed in an in vitro motility assay. The presence of cMyBP-C at an approximate molar ratio of cMyBP-C to whole myosin of 1:2, resulted in a 25% reduction in thin filament velocity (P<0.002) with no effect on relative isometric force under maximally activated conditions (pCa 5). Cardiac MyBP-C was capable of inhibiting actin filament velocity in a concentration-dependent manner using either whole myosin, HMM or S1, indicating that the cMyBP-C does not have to bind to myosin LMM or S2 subdomains to exert its effect. The reduction in velocity by cMyBP-C was independent of changes in ionic strength or excess inorganic phosphate. Cosedimentation experiments demonstrated S1 binding to actin is reduced as a function of cMyBP-C concentration in the presence of ATP. In contrast, S1 avidly bound to actin in the absence of ATP and limited cMyBP-C binding, indicating that cMyBP-C and S1 compete for actin binding in an ATP-dependent fashion. However, based on the relationship between thin filament velocity and filament length, the cMyBP-C induced reduction in velocity was independent of the number of cross-bridges interacting with the thin filament. In conclusion, the effects of cMyBP-C on velocity and force at both maximal and submaximal activation demonstrate that cMyBP-C does not solely act as a tether between the myosin S2 and LMM subdomains but likely affects both the kinetics and recruitment of myosin cross-bridges through its direct interaction with actin and/or myosin head.
Introduction
Cardiac myosin binding protein-C (cMyBP-C) is a thick filament associated sarcomeric protein located in discrete transverse bands in the C zone of the A band. The protein consists of 11 domains (C0–C10), 8 immunoglobulin I-like and 3 fibronectin type 3 motifs, with putative binding of specific domains to the myosin tail (domain C10) [1], titin (domains C8–C10) [2], myosin sub-fragment 2 (S2, domains C1–C2) [3,4], and actin (domains C0–C1) [5,6]. Furthermore, while sedimentation studies indicate that cMyBP-C does not bind to myosin subfragment 1 (S1) [3], the C0–C1 domains may interact with the myosin head and affect contractile function [7,8]. The importance of cMyBP-C in fiber assembly, maintenance of structural integrity, and regulation of contraction is highlighted by the fact that mutations in cMyBP-C are one of the most commonly identified causes of familial hypertrophic cardiomyopathy [9–11].
Studies in muscle fibers where cMyBP-C has been either chemically extracted [12] or transgenically deleted [13] have defined a role of cMyBP-C in decreasing cooperative activation and unloaded shortening speed. This has lead to the hypothesis that cMyBP-C may function as a tether restricting the interaction of myosin with actin. However the mechanism by which cMyBP-C may affect the mechanics and kinetics of myosin as it interacts with actin is currently not well understood. For example, the effect of cMyBP-C on half maximal calcium activated tension (i.e. calcium sensitivity) has been reported to be decreased, unchanged or increased in fibers depending on the experimental approach [12–17].
Fiber studies using N-terminal domain fragments at high stoichiometric concentrations demonstrate that the addition of the C1–C2 or C0–C2 peptide results in a leftward shift in the pCa-tension curve with or without the presence of native cMyBP-C [18–20]. When the C0 domain of MyBP-C was deleted in a transgenic model, again a leftward shift in fiber pCa-tension relation was observed [8]. In contrast, using an expressed C5 domain peptide resulted in a rightward shift, whereas the C0–C1 peptide had no effect on tension [21]. Recent studies in an in vitro motility assay have shown that the cMyBP-C domain fragments may activate the thin filament at low calcium levels but depress thin filament sliding speed at high calcium levels [22]. Thus individual domains appear to have contrasting effects on contractile function making interpretation of the overall function of the intact cMyBP-C challenging.
To delineate the effect of whole cMyBP-C on contractile function we have employed a simplified molecular approach, using purified contractile proteins in the in vitro motility assay. Through this approach we directly characterized the effect of intact cMyBP-C on thin filament activation with respect to velocity of thin filament sliding and isometric force. Moreover, the specific functional significance of cMyBP-C binding to light meromyosin (LMM), myosin S2, and actin was investigated using in vitro motility and cosedimentation assays.
Materials and Methods
Protein Isolation and Characterization
Chicken pectoralis myosin and its subfragments, S1 and HMM, were isolated as previously described [23]. Skeletal myosin was used for it long term stability thus decreasing variability between experiments. Chicken skeletal actin was isolated by standard techniques [24] with further purification obtained through gel filtration chromatography [25]. Beef cardiac troponin and tropomyosin were isolated as previously described [26], with further purification of tropomyosin achieved through hydroxyapatite chromatography. Beef cardiac MyBP-C was isolated per the methods of Hartzell and Glass [27] employing hydroxyapatite and anion exchange chromatography. Regulated thin filaments were reconstituted in a low salt buffer (25 mM KCl, 25 mM imidazole, 5 mM MgCl2, 10 mM DTT and 2 mM EGTA, pH 7.4) with a molar ratio of actin: troponin: tropomyosin of 4:1:1 [28]. Actin and reconstituted thin filaments were labeled with rhodamine-phalloidin (1:1 molar ratio) to allow visualization with epifluorescence microscopy.
As cMyBP-C contains three known phosphorylation sites in the C1–C2 linker [29], the extent to which the isolated cMyBP-C was phosphorylated was determined using the Pro-Q Diamond phosphoprotein gel dye (Invitrogen). Aliquots of cMyBP-C were treated with either protein phosphatase 2A1 (PP2A1, Calbiochem) or protein kinase A (PKA, p2645 Sigma) per previously reported methods [30,31] and compared to untreated cMyBP-C. SDS-PAGE of the treated and untreated cMyBP-C was performed with subsequent Pro-Q Diamond staining per manufacturer’s instructions. The same gel was later stained with Coomassie Blue to confirm similar protein loading between groups. Treatment of cMyBP-C with PKA resulted in a substantial increase in phosphostaining when compared to untreated cMyBP-C (Figure 1). In contrast, treatment of the isolated cMyBP-C with protein phosphatase 2A1 did not result in a reduction in the extent of phosphostaining, indicating that the cMyBP-C used in our experiments was largely dephosphorylated.
Figure 1.
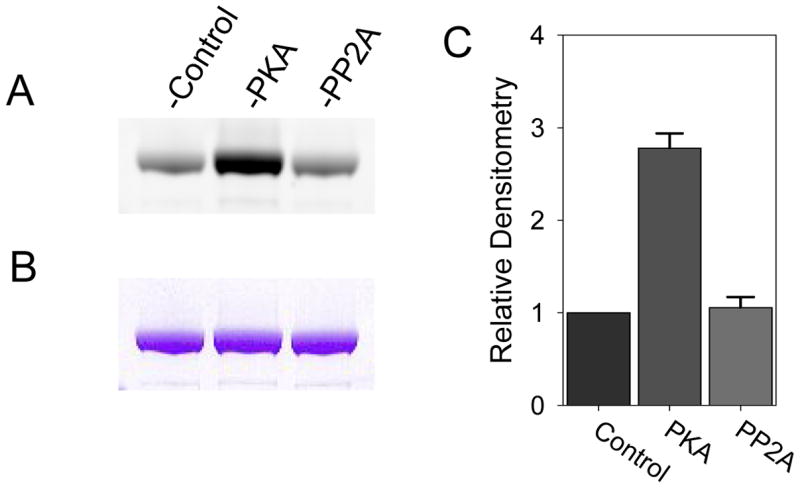
A) ProQ Diamond staining of untreated cMyBP-C (control) and cMyBP-C treated with PKA or PP2A. B) Corresponding Coomassie Blue staining demonstrating similar protein loads for the three groups. C) Densitometry of the ProQ Diamond stain demonstrating increased cMyBP-C phosphorylation with PKA treatment but no decrease in phosphorylation with PP2A treatment indicating that the cMyBP-C used in this study is largely dephosphorylated.
In Vitro Motility Assay
The in vitro motility assay has been previously described in detail by Spudich and colleagues [32]. We have adopted this approach with modification as follows, myosin or its subfragments (100 μg/ml (217 nM for whole myosin)) was applied for one minute to a nitrocellulose coated coverslip unless otherwise specified in 300 mM KCl, 25 mM imidazole, 5 mM MgCl2, 2 mM EGTA and 10 mM DTT. The surface was then washed with bovine serum albumin (0.5 mg/ml) in low salt buffer (25 mM KCl, 25 mM imidazole, 5 mM MgCl2, 2 mM EGTA and 10 mM DTT) to block non-specific protein interactions with the motility surface. Cardiac MyBP-C (cMyBP-C: 100 nM unless otherwise specified) was added to the final motility buffer and compared to controls without cMyBP-C. Employing epifluorescence microscopy, rhodamine-labeled thin filaments were observed moving across the myosin coated surface in the presence of ATP (2 mM) and as a function of the free calcium in the motility solutions (25 mM KCl (unless otherwise specified), 25 mM imidazole, 5 mM MgCl2, 2 mM EGTA and 10 mM DTT, glucose oxidase 0.1 mg/ml, catalase 1.8 μg/ml, glucose 2.3 mg/ml, and 0.38% methylcellulose). Free calcium and ionic strength of the motility solution was determined as previously described [33]. In experiments with actin alone (i.e. no troponin or tropomyosin), the final motility solution is as above with no added calcium. Where noted, KCl and inorganic phosphate (PO4−2) were adjusted in the final motility solutions [28]. These experiments were conducted a minimum of two times. Thin filament motility was recorded on videotape, and subsequently analyzed using the Motion Analysis System VP110 (Motion Analysis Corporation). Typically >300 individual filament velocities were averaged to determine the mean velocity for each motility experiment. The pCa–velocity experiments were conducted three times with the means of individual experiments fit to the Hill equation.
Isometric Force Assay
Relative force was determined by adding increasingly higher concentrations of α-actinin to the motility surface in repeated experiments until thin filament motility was arrested [25]. As α-actinin avidly binds actin, it functions to impede the movement of actin filaments in a concentration dependent manner. To determine isometric force at a single pCa value at least five α-actinin concentrations were used in separate motility experiments to determine the minimum concentration of α-actinin needed to completely arrest thin filament motility. This concentration of α-actinin is a relative measure of isometric force. Force was determined at eight different free calcium concentrations. The pCa–force experiments were conducted twice with the means fit to the Hill equation. For these experiments, α-actinin (Sigma Ltd.) was dialyzed into low salt buffer prior to use. To examine the effects of cMyBP-C on isometric force, 100 nM of cMyBP-C was added to the final motility solution.
Co-Sedimentation Experiments
To determine the effect of cMyBP-C on myosin binding to actin, co-sedimentation experiments were designed where the amount of myosin S1 and cMyBP-C pelleted with actin was determined as a function of cMyBP-C solution concentration. The sedimentation experiments were done in the presence and absence of ATP. This set of experiments was repeated three times. The experiments in which ATP was added, an initial sedimentation spin of equimolar actin and S1 with 5mM ATP was performed (393,000 × g for 20 minutes) and the supernatant was collected. This step removed myosin cross-bridges that strongly bind to actin independent of ATP. Subsequently, increasing concentrations of cMyBP-C (up to 0.6μM) were added to an equimolar mixture of actin and S1 in a high salt buffer (300 mM KCl, 25 mM imidazole, 5 mM MgCl2, 2 mM EGTA and 10 mM DTT). Sedimentation of actin and associated S1 and cMyBP-C was performed through centrifugation at 56 K (137,000 × g) for 20 minutes in the presence and absence of ATP. The pellet was collected and run on SDS-PAGE. Quantification of S1, cMyBP-C and actin in the pellet was performed by densitometric analysis of the gels (Quantity One, Bio-Rad).
Statistical Analysis
The Hill equation was fit to the means of the data [25]. Comparisons between both groups were performed by paired t-test or one-way ANOVA with a Tukey adjustment for multiple comparisons. Data are expressed as mean ± S.E. unless otherwise noted.
Results
Cardiac MyBP-C affects pCa:velocity and force in the motility assay
The addition of cMyBP-C to the motility assay significantly affected thin filament sliding speeds at both maximal and submaximal calcium levels (Figure 2A). Adding 100 nM cMyBP-C at ~ 2:1 molar ratio of whole myosin/cMyBP-C, resulted in a 25% slowing of thin filament velocity at maximal calcium when compared to regulated thin filaments without cMyBP-C (4.6 ± 0.4 μm/s vs. 6.3 ± 0.3 μm/s, respectively, P <0.002). Preincubating cMyBP-C with myosin or adding cMyBP-C prior to adding actin to the motility assay resulted in a similar reduction in thin filament velocity (data not shown). In contrast, cMyBP-C increased thin filament velocity at submaximal calcium levels as demonstrated by a leftward shift in the pCa:velocity relation (pCa50 6.64 ± 0.06 vs. 6.44 ± 0.02, respectively, P = 0.003). The presence of cMyBP-C did not affect the cooperativity of thin filament activation for velocity when compared to control experiments (Hill coefficient 2.4 ± 0.7 vs. 3.6 ± 0.7, respectively, P= 0.18).
Figure 2.
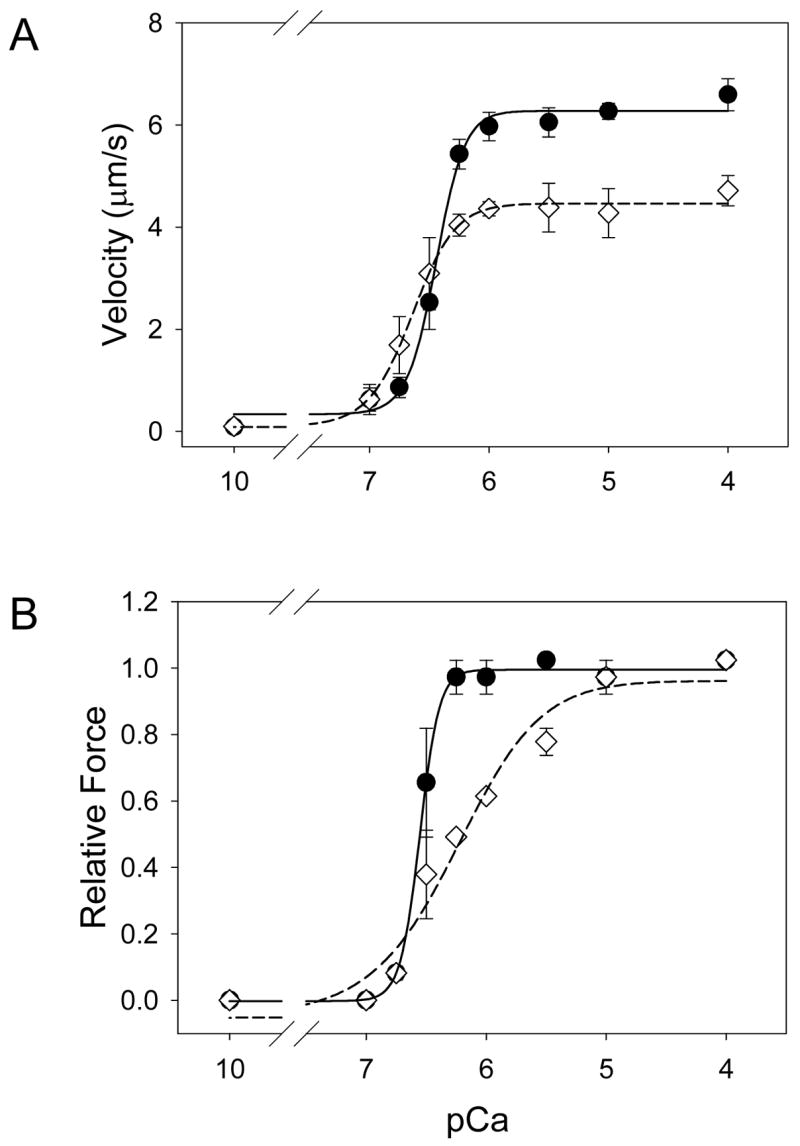
A) pCa-velocity relations for calcium regulated thin filaments in the presence (diamonds, dashed line) and absence of cMyBP-C (circles, solid line). The addition of cMyBP-C to the motility assay resulted in a 25% decrease in maximal velocity and a leftward shift in the pCa:velocity relation (6.64 ± 0.06 vs. control, 6.44 ± 0.03, P=0.003). B) pCa-force relations for calcium regulated thin filaments in the presence (diamonds, dashed line) and absence (circles, solid line) of cMyBP-C. The addition of cMyBP-C had no significant effect on maximal force but caused a rightward shift in the pCa:force relation (6.25 ± 0.09 vs. 6.55 ± 0.02, respectively, P=0.008). Cooperative activation of the thin filament was also reduced in the presence of cMyBP-C as indicated by the Hill coefficients of 1.2 ± 0.3 and. 5.3 ± 1.4, respectively, P=0.005.
Relative isometric force was determined in the motility assay through the use of the actin binding protein, α-actinin (see Methods; Figure 2B). At maximal calcium activation, cMyBP-C had no effect on the relative actomyosin force generation (1.00 ± 0.08 vs. 0.94 ± 0.05, with and without c-MyBP-C, respectively, P=0.64). However, at submaximal calcium, the presence of cMyBP-C resulted in a rightward shift in the pCa:force relation when compared to control experiments (pCa50 6.25 ± 0.09 vs. 6.55 ± 0.02, respectively, P=0.004). The Hill coefficient was more than four fold lower in the presence of cMyBP-C when compared to controls (1.2 ± 0.3 vs. 5.3 ± 1.4, respectively, P = 0.004) demonstrating a negative effect of cMyBP-C on cooperativity. Thus at maximal activation cMyBP-C lowered velocity, while not affecting maximal force, however at submaximal activation, it exerted opposite effects with an increase in thin filament velocity and a decrease in isometric force.
Effect of ionic strength and inorganic phosphate on cMyBP-C function
cMyBP-C may slow thin filament sliding by acting as a load against which the myosin must operate. Such loads are possible if cMyBP-C acts as a tether by binding one end of the molecule to the motility surface, the myosin rod, or myosin S2 and the other end to either the myosin head/neck and/or to the actin filament [29]. To determine the role of this putative effect, we have assumed that the cMyBP-C tether would be non-covalently bound and thus susceptible to changes in ionic strength. In addition, excess inorganic phosphate (Pi) is known to decrease force by limiting the force-generating weak to strong binding cross-bridge transition [34,35] so that Pi can be used to vary the force generated by the myosin population that interacts with the actin filament. We investigated the effect of both ionic strength and elevated Pi on actin filament velocity in the presence and absence of cMyBP-C. Here we assumed that under these experimental conditions that either the load presented by cMyBP-C would be reduced as a function of ionic strength or the force generating capacity of myosin would be reduced as a function of Pi, leading to a change in actin filament velocity as predicted by the force:velocity relation.
Ionic strength was adjusted by varying the KCl concentration in the final motility buffer from 25 mM to 100 mM, corresponding to ionic strengths of 51 mM to 127 mM, respectively. As with regulated thin filaments, the addition of 100 nM cMyBP-C resulted in a lowering of actin filament velocities (Figure 3A). However when the ionic strength of the motility buffers was increased to near physiologic levels no effect on velocity was observed for actin or actin plus cMyBP-C. In addition, 16 mM excess Pi, which would be sufficient to reduce myosin force generation by 50% [35], had no effect on thin filament velocity with or without cMyBP-C (Figure 3B). The ionic strength and Pi experiment results suggest that cMyBP-C does not impart a load on thin filament motility as a result of weak non-specific ionic interactions with either the myosin molecule or actin filament.
Figure 3.
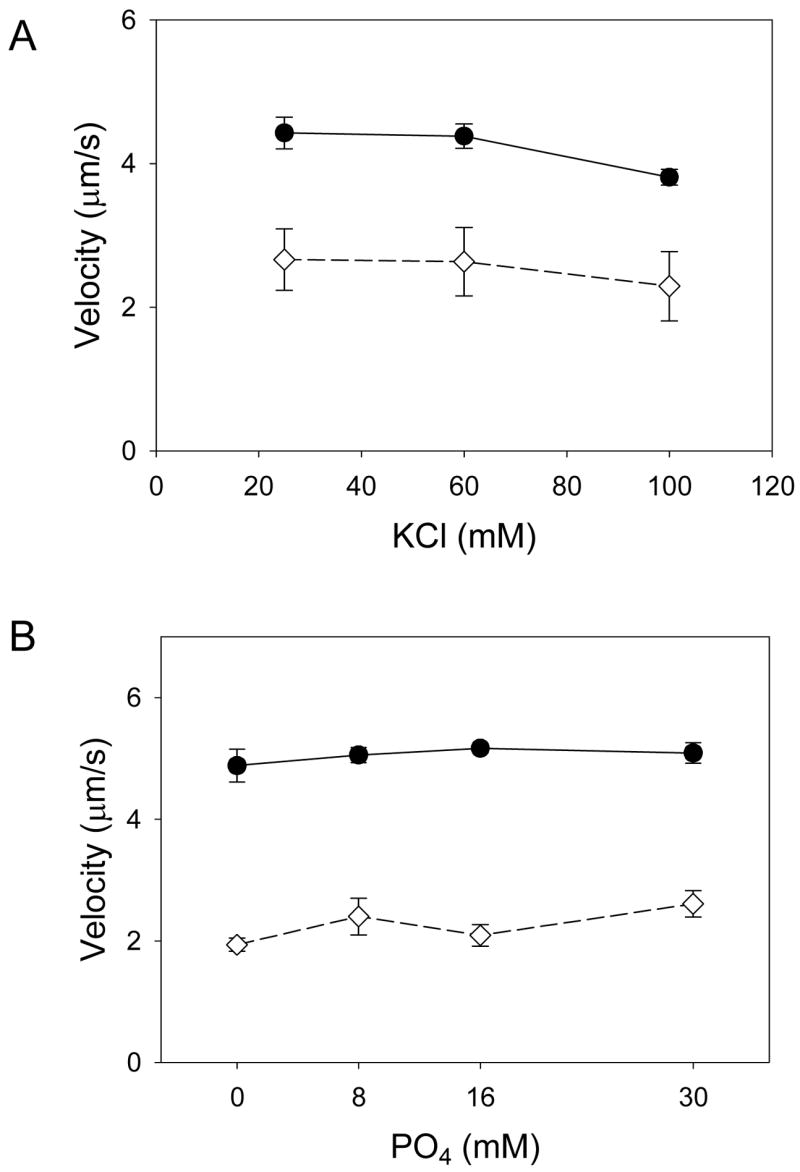
A) Actin filament velocity as a function of ionic strength in the absence (circles, solid line) and presence of cMyBP-C (diamonds, dashed line). No effect of ionic strength was observed indicating that the inhibition of velocity by cMyBP-C was not the result of weak non-specific binding. B) Actin filament velocities as a function of excess inorganic phosphate (Pi) in the absence (circles, solid line) and presence of cMyBP-C (diamonds, dashed line). No effect of excess Pi was observed demonstrating that the reduction in velocity observed cMyBP-C is not the result of cMyBP-C imparting a mechanical load to the system. See text for discussion.
cMyBP-C modulates motility generated by myosin proteolytic fragments
With cMyBP-C capable of binding to the myosin LMM and S2 regions [29], we investigated whether this binding is required for cMyBP-C to induce its functional effect. Accordingly, the effect of cMyBP-C on actin filament velocity generated by whole myosin, HMM and myosin S1 was studied. Cardiac MyBP-C reduced actin filament velocity in a concentration-dependent manner for myosin and the two proteolytic subfragments. For whole myosin and HMM, complete inhibition of actin filament motility occurred at 400 nM cMyBP-C (Figure 4A, B). These experiments were repeated using regulated thin filaments at pCa 5 where similar results were observed (data not shown). Myosin subfragment-1 is capable of sustaining thin filament motility but at much lower velocities than what is observed for either HMM or whole myosin [36]. Similar to the above experiments, myosin S1 actin filament motility was inhibited by cMyBP-C but at lower cMyBP-C concentrations (Figure 4C). These data indicate that the binding of cMyBP-C to LMM or myosin S2 is not required for whole cMyBP-C to exert its inhibitory effect on thin filament motility. Harris and colleagues demonstrated a reduction in thin filament sliding velocity with the C1–C2 domain fragment for both HMM and myosin S1 in the motility assay [22,37]. In these experiments a ten-fold higher concentration of C1–C2 was used to see this effect suggesting that either the binding affinity of this domain fragment is less than intact cMyBP-C protein and/or that whole cMyBP-C affects contractile function in a somewhat different manner than the C1–C2 peptide.
Figure 4.
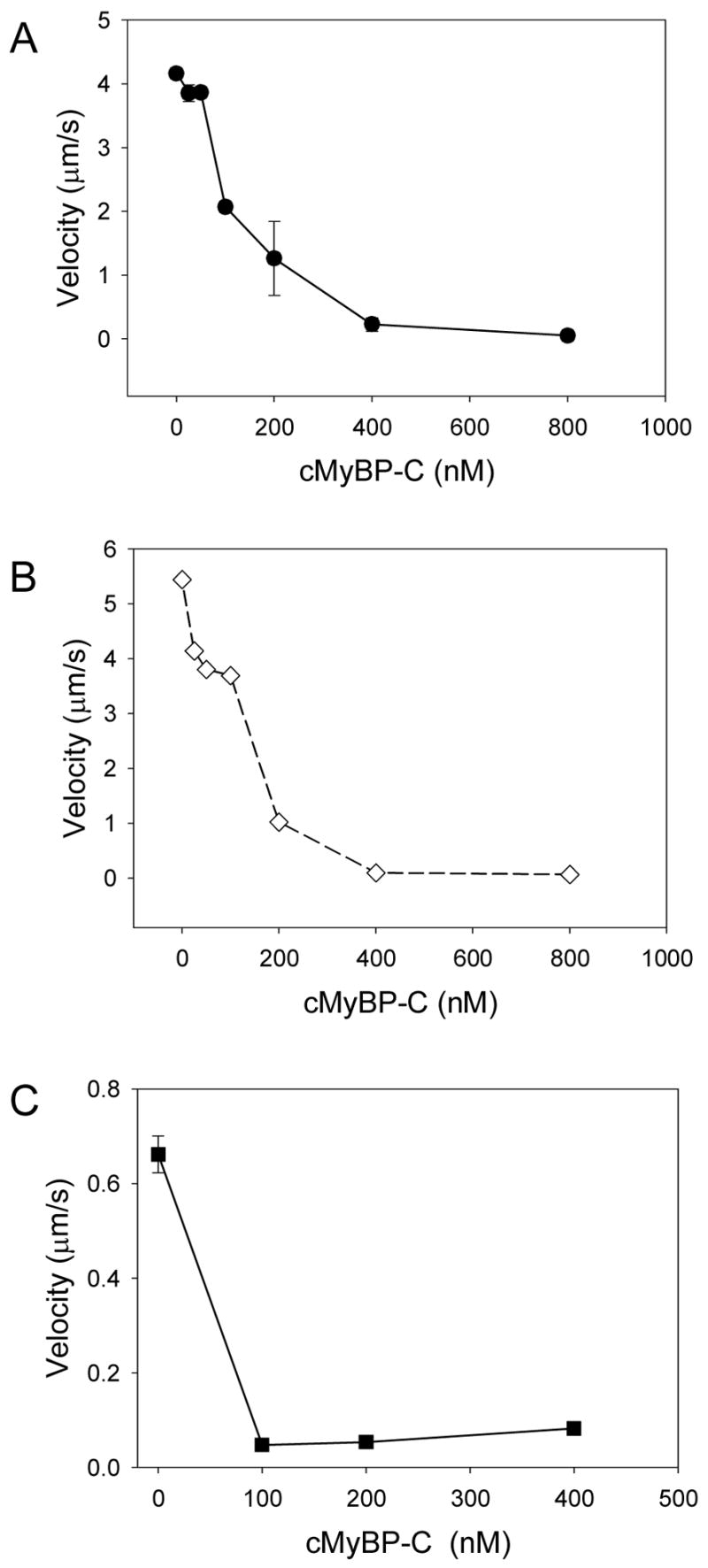
Actin filament velocity as a function of cMyBP-C concentration, using whole myosin (A), HMM (B), and myosin S1 (C). Cardiac MyBP-C caused a concentration dependent reduction in velocity for myosin and its subfragments. These data demonstrate that the inhibition of velocity by cMyBP-C is not the result binding of cMyBP-C LMM or myosin S2.
cMyBP-C is competitive for myosin S1 binding to actin
The potential that cMyBP-C competes with myosin S1 for actin binding was characterized by co-sedimentation experiments. In these experiments cMyBP-C co-sedimented with actin in a concentration dependent manor (Figure 5A, B), and thus demonstrates cMyBP-C’s ability to bind to actin. In contrast, the amount of myosin S1 that binds to actin was reduced as a function of cMyBP-C. These experiments were done in the presence of ATP where myosin weakly binds to actin [38]. Next, to determine if cMyBP-C can also affect myosin strong binding to actin (i.e. rigor), experiments were performed in the absence of ATP (Figure 5C, D). Under these conditions, cMyBP-C did not affect myosin S1 binding to actin. Moreover, the amount of cMyBP-C that co-sedimented with actin was reduced nearly 3-fold. These data indicate that cMyBP-C and myosin compete for actin binding but the extent of competition is dependent on the myosin binding state.
Figure 5.

Co-sedimentation of myosin S1 and cMyBP-C with actin. Representative SDS PAGE (A) and composite densitometric ratios (B) of cMyBP-C (circles) and myosin S1 (squares) cosedimented with actin in the presence of ATP. As is demonstrated the amount of myosin S1 cosedimented with actin is reduced with the addition of cMyBP-C. In contrast, in experiments where ATP was not added (C and D), myosin rigor binding to actin is not affect by cMyBP-C but cMyBP-C binding to actin is reduced nearly 3 fold. These data demonstrate that cMyBP-C competes with myosin S1 for actin binding.
cMyBP-C alters actomyosin kinetics
The reduction in thin filament velocity by cMyBP-C could be due to cMyBP-C limiting the number of myosin cross-bridges interacting with the thin filament or that cMyBP-C directly affects the mechanokinetics of actomyosin. The cosedimentation experiment indicates that cMyBP-C is capable of limiting myosin binding to actin and could in part affect thin filament motility. Thin filament velocity is limited by the number of cross-bridges that interact with the actin filament, reaching an asymptote at high cross-bridge numbers (i.e. >20 for muscle myosins having ~5% duty ratios [39]). This was observed in Figure 6 where thin filament velocities at pCa 5 increase as a function of thin filament length, given that longer filaments can interact with a greater number of cross-bridges on the motility surface. If it assumed that cMyBP-C slows thin filament velocities solely by limiting the number of cross-bridges that can interact with the thin filament, then velocities for the longest thin filaments should approach the asymptote observed in the absence of cMyBP-C (see legend for Figure 6). This was not the case, suggesting that a more likely scenario is that cMyBP-C directly modulates the cross-bridge detachment rate, which is the rate limiting step for filament sliding velocity when cross-bridge number is not limiting [40].
Figure 6.
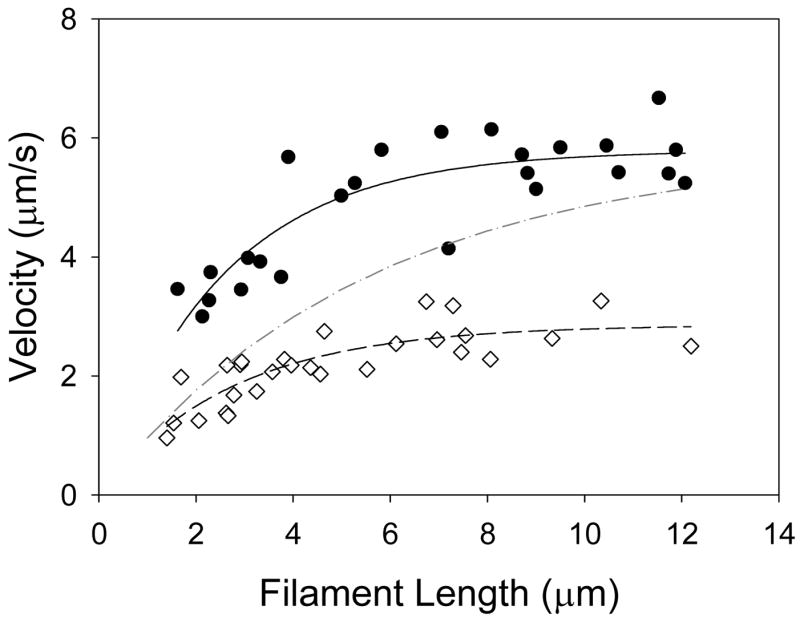
Thin filament velocity of individual thin filaments as a function of thin filament length with (diamonds, dashed line) and without (circles, solid line) cMyBP-C using a sparsely populated myosin-coated motility surface (myosin loading buffer concentration of 25μg/ml). The number of myosin cross-bridges interacting with the thin filament is a function of its length. This relation can be expressed mathematically [39]: v = vmax[1−(1−fxb)n], where v is the filament velocity, vmax is the maximum filament velocity, fxb is the cross-bridge duty ratio, and n is the number of cross-bridges available to interact with the thin filament. For the purposes of the fit we assume that the duty ratio was 5% [39]. Accordingly, at short filament lengths (e.g. <6μm) thin filament velocity is dependent on the number of cross-bridges with which it interacts (i.e. filament length). At longer filament lengths (e.g. >6μm) thin filament velocity is independent of filament length. As depicted in this figure with filaments > 6μm, cMyBP-C caused a reduction in thin filament velocity. If cross-bridge number was the sole determinant of the cMyBP-C reduction in velocity at long filament lengths, then velocity should recover to that observed in the absence of cMyBP-C, which is not the case. This is hypothetically represented by the dash-dot curve where it was assumed that the number of cross-bridges interacting with the thin filament is reduced by 50% and vmax is unchanged with the addition of cMyBP-C.
Discussion
In this study we used the in vitro motility assay, a molecular model system for muscle contraction, to define the mechanism by which cMyBP-C modulates actomyosin function. An advantage of this approach is that the effect of cMyBP-C on regulated thin filament motility generated by myosin and its proteolytic fragments can be studied at any desired stoichiometry. With the use of monodispersed myosin molecules devoid of thick filament associated proteins most notably titin, we demonstrate that cMyBP-C does not require an organized filament lattice to exert its effects on force and velocity. Moreover whole cMyBP-C is able to reduce thin filament velocity and myosin binding to actin independent of binding to LMM and myosin S2. These data in conjunction with the ionic strength and inorganic phosphate experiments suggest that cMyBP-C is capable of exerting its effects on thin filament motility independent of a putative tether-like mechanism involving the simultaneous binding of cMyBP-C to LMM and myosin S2. A similar conclusion was drawn by Harris et al. [19] studying the effect of exogenous C1-C2 peptide fragments in muscle fibers. Given that cMyBP-C is capable of binding to multiple proteins within the sarcomere it is conceivable that cMyBP-C can bind to two proteins or locations simultaneously thus creating a tether-like link. However, based on our results, we propose that cMyBP-C can affect muscle contractility by directly modulating actomyosin binding and kinetics. Thus, these data suggest that cMyBP-C binding to the thin filament may play a role in the modulatory capacity of cMyBP-C.
At maximal calcium activation, cMyBP-C caused a decrease in the sliding velocity of regulated thin filaments when compared to control (Figure 2A). These results are consistent with previous published experiments in muscle fibers where an increase in the unloaded shortening velocity was demonstrated with either chemical extraction [41] or transgenic deletion of cMyBP-C [13,42]. Compared to the skinned fiber studies, the motility assay allowed us to characterize the effect of cMyBP-C on unloaded shortening over the full physiologic calcium concentration range. In contrast to the decrease in velocity seen at high calcium levels, we demonstrate an increase in velocity with cMyBP-C at submaximal calcium concentrations. While the molar ratio of cMyBP-C to whole myosin (1:2) used in these experiments is close to that observed in the C-zone of the myofilament (1:3), the magnitude of the cMyBP-C effects on velocity may be accentuated in this study when compared to intact muscle where the molar ratio of cMyBP-C to myosin is 1:7.
Several investigators have proposed that the reduced unloaded shortening velocity in fiber studies is the result of cMyBP-C functioning either as a tether restricting myosin’s interactions with actin [15,29,43–45] and/or imparting a viscous load to thick-thin filament sliding [46]. Our data would indicate that neither of these mechanisms can account for the functional effects observed with cMyBP-C in this study. For example, cMyBP-C could hypothetically tether the myosin head/neck or actin filament to the motility surface via cMyBP-C non-specifically binding to the motility surface. This is unlikely since the motility surface is blocked with bovine serum albumin prior to the addition of cMyBP-C. However, cMyBP-C does bind in vitro to both the myosin LMM and S2 segments [3], providing a more physiologically relevant point of attachment. The fact that cMyBP-C can reduce actin filament velocities independent of LMM and myosin S2 binding (using HMM and myosin S1, respectively; see Figure 4) indicates that the reduction in velocity is not likely the result of a tether involving the binding of cMyBP- C to LMM or myosin S2. Moreover, if thin filaments were moving under a load due to the presence of cMyBP-C, then changing the ratio of force generating myosin molecules to cMyBP-C should have led to significant changes in thin filament velocity, which was not observed when excess Pi [35] was used in the motility assay (Figure 3B). Finally, an internal load created by cMyBP-C does not appear to exist in the motility assay where an increase rather than a decrease in thin filament velocity was observed at low calcium levels with the addition of cMyBP-C compared to controls. Moreover, in the force experiments a similar concentration of α-actinin was used to arrest thin filament motility at high calcium concentrations which would not be anticipated if cMyBP-C was imposing a load on the system.
Alternatively, it has been proposed that cMyBP-C may bind directly to actin [6] and through this interaction affect contractile function. Since myosin S1 does not bind cMyBP-C [3], the inhibition of actin velocities observed using myosin S1 would suggest that this effect is mediated via cMyBP-C binding to actin. In fact, the co-sedimentation experiments demonstrate that cMyBP-C and myosin compete for actin binding as a function of ATP (Figure 5). However, a reduced number of cross-bridges interacting with actin cannot solely account for the cMyBP-C induced reduction in velocity, as long actin filaments, which interact with greater number of cross-bridges on the motility surface, do not exhibit faster velocities (see Results; Figure 6). Thus, cMyBP-C, presumably through its interaction with the thin filament, modulates either the inherent mechanics and/or kinetics of the myosin cross-bridge [22]. Specifically, cMyBP-C could reduce the displacement generated by the myosin motor and/or the rate at which the cross-bridge detaches from actin following the powerstroke, both of which govern actin filament velocity at the molecular level [47]. Evidence for a kinetic effect already exists given that MyBP-C slows the rate of actomyosin ATP hydrolysis [48,49] and is consistent with a slower cross-bridge detachment rate being the most likely effect of cMyBP-C on thin filament velocity.
No change in maximal isometric force is observed with the addition of cMyBP-C, which is in agreement with results from fiber studies where cMyBP-C is either extracted [41] or transgenically knocked out [42]. If cMyBP-C decreases the rate of cross-bridge detachment as suggested above this would result in an increased cross-bridge attachment time [47]. A determinant of isometric force is the duty ratio, defined as the myosin attachment time divided by the total cycle time (i.e., time of the ATP hydrolysis cycle). As maximal isometric force is unaffected by cMyBP-C, this would suggest that the ATP hydrolysis rate is likely slowed [5] accordingly so that the duty ratio is preserved in the presence of cMyBP-C. Why this is not the case at submaximal calcium levels where force is reduced in the presence of cMyBP-C is a matter of speculation. As crossbridge kinetics are differentially affected by load and the degree of calcium activation [50], these data would suggest that cross-bridge number and kinetics are likely responsible for the differential effect of cMyBP-C on both force and velocity at submaximal calcium.
In conclusion, these motility studies demonstrate that cMyBP-C can exert its modulatory role on actomyosin force and motion generation without the spatial constraints that normally exist within the sarcomere. While the effects seen in the motility assay may not completely reflect the binding and function of cMyBP-C in the intact lattice, where structural organization and the presence of other sarcomere proteins may affect contractile performance, we have demonstrated that cMyBP-C elicits the same effect on calcium-activated force and velocity as previously demonstrated in muscle fibers. Within the sarcomere, the cMyBP-C to myosin molar ratio of 1:7 and its location being limited to the C zone suggests that cMyBP-C exerts its effect either directly on those cross-bridges with which it interacts or that its effects are cooperative and must be communicated to other cross-bridges within the thick filament either through the thick filament backbone or through the thin filament. Distinguishing between these two possibilities will be difficult based on fiber studies, whereas the in vitro approaches described here in combination with native thick filaments that maintain the in vivo stoichiometry and spatial relationships may provide an answer to this important question [51–53]. Presently, with the use of monodispersed myosin at a 2:1 whole myosin/cMyBP-C molar ratio, we have demonstrated that cMyBP-C directly affects actomyosin kinetics with the effect being independent of cMyBP-C binding to the LMM and S2 domains of myosin. These results raise the possibility that cMyBP-C exerts its modulatory role via its interaction with actin, which may be a regulated property depending on cMyBP-C’s phosphorylation state [6]. It should be noted that neither the specific cMyBP-C binding site on actin nor the physiologic relevance of such binding in muscle fibers (if present) has been defined. Thus, future studies characterizing the effect of cMyBP-C on the inherent mechanics and kinetics of the individual cross-bridge in the laser trap [54] using intact native thick filament [51] should offer insight into the molecular mechanisms by which cMyBP-C exerts its effect on contractility and its regulation by cMyBP-C phosphorylation.
Acknowledgments
This work was supported NIH grants R01 HL77637 (PVB) and P01 HL59408 (DMW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol. 1993;123:619–26. doi: 10.1083/jcb.123.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freiburg A, Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem. 1996;235:317–23. doi: 10.1111/j.1432-1033.1996.00317.x. [DOI] [PubMed] [Google Scholar]
- 3.Starr R, Offer G. The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem J. 1978;171:813–6. doi: 10.1042/bj1710813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–49. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 5.Moos C, Mason CM, Besterman JM, Feng IN, Dubin JH. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1 1. J Mol Biol. 1978;124:571–86. doi: 10.1016/0022-2836(78)90172-9. [DOI] [PubMed] [Google Scholar]
- 6.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol. 2003;331:713–24. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 7.Flavigny J, Souchet M, Sebillon P, Berrebi-Bertrand I, Hainque B, Mallet A, Bril A, Schwartz K, Carrier L. COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. Journal of Molecular Biology. 1999;294:443–56. doi: 10.1006/jmbi.1999.3276. [DOI] [PubMed] [Google Scholar]
- 8.Witt CC, Gerull B, Davies MJ, Centner T, Linke WA, Thierfelder L. Hypercontractile Properties of Cardiac Muscle Fibers in a Knock-in Mouse Model of Cardiac Myosin-binding Protein-C. J Biol Chem. 2001;276:5353–9. doi: 10.1074/jbc.M008691200. [DOI] [PubMed] [Google Scholar]
- 9.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–40. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 11.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J Gen Physiol. 1991;97:1141–63. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer BM, Georgakopoulos D, Janssen PM, Wang Y, Alpert NR, Belardi DF, Harris SP, Moss RL, Burgon PG, Seidman CE, Seidman JG, Maughan DW, Kass DA. Role of Cardiac Myosin Binding Protein C in Sustaining Left Ventricular Systolic Stiffening. Circ Res. 2004;94:1249–55. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 14.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys J. 2006;90:4119–27. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 16.Kulikovskaya I, McClellan G, Levine R, Winegrad S. Effect of extraction of myosin binding protein C on contractility of rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H857–H865. doi: 10.1152/ajpheart.00841.2002. [DOI] [PubMed] [Google Scholar]
- 17.Palmer BM, McConnell BK, Li GH, Seidman CE, Seidman JG, Irving TC, Alpert NR, Maughan DW. Reduced cross-bridge dependent stiffness of skinned myocardium from mice lacking cardiac myosin binding protein-C. Mol Cell Biochem. 2004;263:73–80. doi: 10.1023/B:MCBI.0000041849.60591.45. [DOI] [PubMed] [Google Scholar]
- 18.Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin Binding Protein C, a Phosphorylation-Dependent Force Regulator in Muscle That Controls the Attachment of Myosin Heads by Its Interaction With Myosin S2. Circ Res. 2000;86:51–8. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 19.Harris SP, Rostkova E, Gautel M, Moss RL. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism 1. Circ Res. 2004;95:930–6. doi: 10.1161/01.RES.0000147312.02673.56. [DOI] [PubMed] [Google Scholar]
- 20.Herron TJ, Rostkova E, Kunst G, Chaturvedi R, Gautel M, Kentish JC. Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ Res. 2006;98:1290–8. doi: 10.1161/01.RES.0000222059.54917.ef. [DOI] [PubMed] [Google Scholar]
- 21.McClellan G, Kulikovskaya I, Flavigny J, Carrier L, Winegrad S. Effect of cardiac myosin-binding protein C on stability of the thick filament. Journal of Molecular and Cellular Cardiology. 2004;37:823–35. doi: 10.1016/j.yjmcc.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 22.Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846–54. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 23.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982;85(Pt B):55–71. 55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 24.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B):164–81. 164–81. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 25.VanBuren P, Alix SL, Gorga JA, Begin KJ, LeWinter MM, Alpert NR. Cardiac troponin T isoforms demonstrate similar effects on mechanical performance in a regulated contractile system. Am J Physiol Heart Circ Physiol. 2002;282:H1665–H1671. doi: 10.1152/ajpheart.00938.2001. [DOI] [PubMed] [Google Scholar]
- 26.Potter JD. Preparation of troponin and its subunits. Methods Enzymol. 1982;85(Pt B):241–63. 241–63. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- 27.Hartzell HC, Glass DB. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem. 1984;259:15587–96. [PubMed] [Google Scholar]
- 28.Gorga JA, Fishbaugher DE, VanBuren P. Activation of the calcium-regulated thin filament by myosin strong binding. Biophys J. 2003;85:2484–91. doi: 10.1016/s0006-3495(03)74671-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: its role in physiology and disease 1. Circ Res. 2004;94:1279–89. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 30.Hunlich M, Begin KJ, Gorga JA, Fishbaugher DE, LeWinter MM, VanBuren P. Protein kinase A mediated modulation of acto-myosin kinetics. J Mol Cell Cardiol. 2005;38:119–25. doi: 10.1016/j.yjmcc.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Noguchi T, Hunlich M, Camp PC, Begin KJ, El Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Thin filament-based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–7. doi: 10.1161/01.CIR.0000139334.43109.F9. [DOI] [PubMed] [Google Scholar]
- 32.Kron SJ, Toyoshima YY, Uyeda TQ, Spudich JA. Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 1991;196:399–416. 399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 33.Brooks SP, Storey KB. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal Biochem. 1992;201:119–26. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 34.Gordon AM, Regnier M, Homsher E. Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls”. News Physiol Sci. 2001;16:49–55. [PubMed] [Google Scholar]
- 35.Millar NC, Homsher E. The effect of phosphate and calcium on force generation in glycerinated rabbit skeletal muscle fibers. A steady-state and transient kinetic study. J Biol Chem. 1990;265:20234–40. [PubMed] [Google Scholar]
- 36.Toyoshima YY, Kron SJ, McNally EM, Niebling KR, Toyoshima C, Spudich JA. Myosin subfragment-1 is sufficient to move actin filaments in vitro. Nature. 1987;328:536–9. doi: 10.1038/328536a0. [DOI] [PubMed] [Google Scholar]
- 37.Shaffer JF, Razumova MV, Tu AY, Regnier M, Harris SP. Myosin S2 is not required for effects of myosin binding protein-C on motility. FEBS Lett. 2007;581:1501–4. doi: 10.1016/j.febslet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Eisenberg E, Hill TL. A cross-bridge model of muscle contraction. Prog Biophys Mol Biol. 1978;33:55–82. doi: 10.1016/0079-6107(79)90025-7. [DOI] [PubMed] [Google Scholar]
- 39.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem. 1993;268:14764–8. [PubMed] [Google Scholar]
- 40.Siemankowski RF, Wiseman MO, White HD. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci U S A. 1985;82:658–62. doi: 10.1073/pnas.82.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991;439:701–15. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded Shortening, Power Output, and Rate of Force Redevelopment Are Increased With Knockout of Cardiac Myosin Binding Protein-C. Circ Res. 2003;93:752–8. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 43.Calaghan SC, Trinick J, Knight PJ, White E. A role for C-protein in the regulation of contraction and intracellular Ca2+ in intact rat ventricular myocytes. J Physiol (Lond) 2000;528:151–6. doi: 10.1111/j.1469-7793.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci U S A. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winegrad S. Cardiac Myosin Binding Protein C. Circ Res. 1999;84:1117–26. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 46.Palmer BM, Noguchi T, Wang Y, Heim JR, Alpert NR, Burgon PG, Seidman CE, Seidman JG, Maughan DW, LeWinter MM. Effect of Cardiac Myosin Binding Protein-C on Mechanoenergetics in Mouse Myocardium. Circ Res. 2004;94:1615–22. doi: 10.1161/01.RES.0000132744.08754.f2. [DOI] [PubMed] [Google Scholar]
- 47.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 48.Moos C, Feng IN. Effect of C-protein on actomyosin ATPase. Biochim Biophys Acta. 1980;632:141–9. doi: 10.1016/0304-4165(80)90071-9. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto K, Moos C. The C-proteins of rabbit red, white, and cardiac muscles. J Biol Chem. 1983;258:8395–401. [PubMed] [Google Scholar]
- 50.Kad NM, Kim S, Warshaw DM, VanBuren P, Baker JE. Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. PNAS. 2005;102:16990–5. doi: 10.1073/pnas.0506326102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine R, Weisberg A, Kulikovskaya I, McClellan G, Winegrad S. Multiple structures of thick filaments in resting cardiac muscle and their influence on cross-bridge interactions. Biophys J. 2001;81:1070–82. doi: 10.1016/S0006-3495(01)75764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kensler RW, Harris SP. The Structure of Isolated Cardiac Myosin Thick Filaments from cMyBP-C Knockout Mice. Biophys J. 2008;94:1707–18. doi: 10.1529/biophysj.107.115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guilford WH, Dupuis DE, Kennedy G, Wu J, Patlak JB, Warshaw DM. Smooth muscle and skeletal muscle myosins produce similar unitary forces and displacements in the laser trap. Biophys J. 1997;72:1006–21. doi: 10.1016/S0006-3495(97)78753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]