

Abstract
Infection by human papillomavirus (HPV) is a major risk factor for human cervical carcinoma. However, the HPV infection alone is not sufficient for cancer formation. Cervical carcinogenesis is considered a multistep process accompanied by genetic alterations of the cell. Ras is activated in approximately 20% of human cancers, and it is related to the metastatic conversion of tumor cells. We investigated how Ras activation was involved in the malignant conversion of HPV-infected lesions. The active form of H-ras was introduced into human primary keratinocytes expressing the HPV type 18 (HPV18) oncoproteins E6 and/or E7. We analyzed the keratinocytes’ growth potentials and found that the activation of the Ras pathway induced senescence-like growth arrest. Senescence could be eliminated by high-risk E7 expression, suggesting that the pRb pathway was important for Ras-induced senescence. Then we analyzed the effect of Ras activation on epidermis development by using an organotypic “raft” culture and found that the E7 and H-ras coexpressions conferred invasive potential on the epidermis. This invasiveness resulted from the upregulation of MT1-MMP and MMP9 by H-ras and E7, respectively, in which the activation of the MEK/extracellular signal-regulated kinase pathway was involved. These results indicated that the activation of Ras or the related signal pathways promoted the malignant conversion of HPV-infected cells.
It has been proposed that carcinogenesis is a multistep process accompanied by multiple genetic alterations (4, 55). Hyperplasia is considered the first step of carcinogenesis, which is induced by inflammation, activation of growth signals, or organ regeneration. Among the expanding population of cells, some cells acquire genetic mutations in the genes that regulate cell division, motility, and invasiveness. If the mutation disrupts the cell cycle checkpoint machinery, genetic instability is induced, resulting in the acceleration of genetic alterations. Immortalization can be induced by reactivation of telomerase and/or disruption of the pRb pathway. These multiple processes are required for the malignant conversion of the cell.
Cervical cancer is one of the gynecological cancers. Human papillomaviruses (HPVs) are detected in more than 90% of cervical cancer cases (59), and HPV infection is considered a major causative factor for the cancer. HPVs are small DNA viruses which have about 8,000-bp, double-strand DNA as the genome (25). More than 100 types of HPVs have been reported (59), which are classified as low-risk and high-risk types according to their associations with malignant tumors (59). High-risk HPVs encode two oncogenes, E6 and E7, which play important roles in carcinogenesis (36). E6 has two zinc finger domains and interacts with tumor suppressor p53 (7) and degrades it to escape from apoptosis and to disrupt cell cycle checkpoint machinery (45). E7 interacts with the tumor suppressor pRb and degrades or inactivates it to escape from the checkpoint machinery and to induce genome instability (11, 12). Although the inactivation of both p53 and pRb seems to be pivotal for oncogenesis by HPV, other functions of these oncoproteins also contribute to carcinogenesis. E6 is able to reactivate human telomerase activity, resulting in immortalization of the infected cell (29). E6 has a PDZ-binding domain at the C terminus that interacts with PDZ proteins (32), such as hDlg and hScrib, the association of which is reported to be involved in the transformation function (28, 38). E7 inactivates Cdk inhibitor p21 and disrupts the cell cycle checkpoint (23, 27) and enhances myc expression to upregulate cell proliferation (40, 41).
Although HPV infection is critical for cervical oncogenesis, most infected lesions do not proceed to the malignant tumor stage, and a long incubation period is required for cancer development (14, 46), indicating that HPV infection alone is not sufficient and that several genetic alterations of the cell are required for the induction of cervical cancer. Even though many mutations have been reported for HPV-positive cervical cancer (54), the critical genes responsible for cancer development have not been identified.
ras is a proto-oncogene which is frequently mutated or amplified in human cancers (3). Activating types of ras mutations are detected in approximately 20% of all human cancers (10). Ras regulates cell proliferation and migration through the activation of mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase, and the Rac/Rho signaling pathways (48). ras mutation and amplification have also been detected in some cases of cervical cancers, and the involvement of Ras activation in HPV-induced carcinogenesis was proposed by several reports (2, 44). Ras activation upregulated the gene expression levels of E6 and E7 (34). Ras was reported to contribute to cellular transformation in cooperation with E6 and E7 in the mouse model (19, 47). With clinical samples, the rate of ras mutation was reported to increase in association with the lesion's grade of cervical neoplasia (2). These findings suggested that Ras activation is one of the major genetic alterations in the development of cervical cancer.
To clarify HPV-induced carcinogenesis and to prevent cancer progression, an understanding of the HPV life cycle is essential. It is known that HPV replication has an intimate connection with the differentiation process of keratinocytes (39, 56). Virion production is restricted in the differentiated layer of epidermis; therefore, the standard monolayer culture of keratinocytes is unable to support the HPV life cycle. To analyze virion production, a culture medium containing a high concentration of calcium ions (15) or a suspension culture with semisolid medium (43) has been employed, by which some aspects of viral replication have been studied. An organotypic raft culture is one of the culture systems used for studying viral replication, which can reproduce an epidermal structure in vitro and support the whole life cycle of HPV (9, 35). It seems better to use the culture condition that can support HPV replication for evaluating viral gene functions and for analyzing virus-induced oncogenesis because some of these aspects might be cell and/or tissue specific.
In this report, we analyzed the effects of Ras activation on HPV-induced tumorigenesis, using human primary keratinocytes. Studies of the transformation activity of Ras were performed mainly with rodent fibroblasts and mouse model systems. As noted above, however, it is important to use the assay system suitable for studying the HPV life cycle. We employed the organotypic raft culture and found functions of E7 and Ras that cooperated in the acquisition of invasiveness. We also found that the induction of matrix metalloproteases (MMPs) by E7 and H-ras was involved in invasiveness. These observations indicated that the activation of Ras or the related pathways conferred invasive potential on HPV-infected cells.
MATERIALS AND METHODS
Vector constructions.
HPV oncogenes (E6 and E7) were isolated by PCR with a full-length HPV type 18 (HPV18) clone (GenBank accession number X05015) as the template and then inserted into the pLXSN (BD Biosciences Clontech, Palo Alto, CA; GenBank accession number M28248) vector. The active form of the human H-ras (G12V) gene, which was inserted into the pMXpuro vector with a hemagglutinin tag at the N terminus, was provided by Ishikawa (Laboratory of Cell Cycle Regulation, Dept. of Gene Mechanisms, Div. of Integrated Life Science, Graduate School of Biostudies, Kyoto University, Japan). The DNA fragment containing the H-ras sequence was isolated by EcoRI and then cut by BglII. The fragment was inserted into the pLPCX (BD Biosciences Clontech, Palo Alto, CA) vector. MAPK constitutive active mutants (mek1-SDSE [Xenopus, S218D, S222E], mkk7-DED [mouse, S287D, T291E, S293D], and mkk6-SETE [human, S207E, T211E]) (21) were provided by Nishida (Laboratory of Signal Transduction, Dept. of Cell and Developmental Biology, Div. of Integrated Life Science, Graduate School of Biostudies, Kyoto University, Japan). The DNAs encoding these MAPK mutants were amplified by PCR and then inserted into pLPCX. The short hairpin RNA (shRNA) sequences used for pRb were GAT ACT AGA TCG TGT CAG ATT CAA GAG ATC TGA CAT GAT CTG GTA TCT TTT TTA TCG AT. The annealed oligonucleotides were inserted into the pSIREN-RetroQ vector (BD Biosciences Clontech, Palo Alto, CA).
Cell cultures.
Human foreskin keratinocytes (HFKs; Kurabo Industries, Ltd., Osaka, Japan) were maintained with serum-free keratinocyte growth medium (KGM; product, EpiLife-KG2; Kurabo). Human foreskin fibroblasts (HFFs; Kurabo), HeLa cells, and 293T cells were maintained with Dulbecco's modified minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (growth medium).
Retroviral production and infection.
293T cells were transfected with the retroviral gene expression plasmid (3.5 μg; pLXSN or pLPCX), pCL10A1 (1 μg; Imgenex Corp., San Diego, CA), and pGreenLantern-1 (0.5 μg; Invitrogen Corp., Carlsbad, CA) and herring sperm DNA (5 μg; Roche Diagnostics, GmbH, Mannheim, Germany) by using the calcium phosphate coprecipitation method. At day 2 after transfection, the culture medium was replaced with fresh KGM, and the retroviral vector was collected in the medium for 8 h. The conditioned medium containing viral vector was clarified through a 0.45-μm-pore filter and used for the infection of HFKs. For the infection procedure, the previous KGM was removed, and the virus-containing medium mixed with 8 μg/ml polybrene was overlaid on the HFKs. The cells were maintained for 6 h in a CO2 incubator, and then the medium was changed to fresh KGM. At 24 h after infection, the cells were spread at a low density. G418 (60 μg/ml) and puromycin (0.2 μg/ml) were added to the medium at 48 h after infection, and the cells were incubated for 4 to 7 days.
SA-β-gal assay.
H-ras and HPV oncoprotein-coexpressing HFKs were spread at 1.0 × 105 cells/dish. After 24 h, an evaluation of senescence was performed with a senescence-associated β-galactosidase (SA-β-gal) assay kit (Cell Signaling Technology, Inc., Danvers, MA) by following the manufacture's instructions.
Immunoblot analysis.
Total cell lysates were prepared with triple-detergent lysis buffer (50 mM Tris HCl [pH 8.0], 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40, 0.5% sodium deoxycholate) supplemented with protease inhibitor cocktail (Nakarai Tesque, Kyoto, Japan). The cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, and immunoblot analysis was performed by using a standard protocol. The antibodies used in the experiment were anti-18-E7 (clone C-20), anti-H-Ras (clone 259) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and an pRb-specific antibody (BD Biosciences Pharmingen, San Diego, CA).
Organotypic raft culture.
For a preparation of the dermal equivalent, one part type I collagen (cell matrix type I-P; Nitta Gelatin Co., Ltd., Osaka, Japan) and two parts growth medium containing HFFs (1 × 106 cells) were mixed while they cooled and were poured into a 6-cm dish, and then the cells were maintained in a 5% CO2 incubator at 37°C until the collagen gel contracted to about a 2-cm diameter. The gel was soaked in fresh KGM for several hours and then transferred in a Transwell insert. The gel was put in a 6-well plate, and fresh KGM was filled into both the bottom and the insert. HFKs were overlaid on the gel (day 0). At day 1, the medium was changed to a mixture of KGM and DMEM growth medium (KGM/DMEM, 1:1), and 24 h later, the medium was changed to the same mixture, adjusting the CaCl2 concentration to 1.8 mM (day 2). On day 3, the surface of the collagen gel was exposed to air, and then the medium in the bottom was changed to fresh medium every day. Multilayered cultures of keratinocytes were obtained at day 10.
The construction of the raft culture with HeLa cells was performed by a protocol similar to that described above, except that DMEM supplemented with 10% fetal bovine serum was used for the HeLa cells.
Hematoxylin and eosin (H&E) staining.
Specimens of raft culture were embedded in optimal cutting temperature compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan), and thin sections (7 μm) were obtained by cryosectioning (Cryo-Star HM560M machine; Microm Laborgeräte GmbH, Walldrof, Germany). The sections were transferred onto slide glasses and dried. They were stained with hematoxylin 3G (Sakura) for 5 min. After sections were washed with water, they were rinsed with HCl (4 × 10−3 N) for about 1 min and washed again. They were then stained with a pure eosin solution (Sakura) for 3 min and destained with 80% ethanol.
Immunohistochemical analysis.
The cryosections were fixed with Mildform 10N (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and then treated with a target retrieval solution (Dako Japan KK, Kyoto, Japan). The endogenous peroxidase activity was quenched with 0.3% H2O2-MeOH after the target retrieval process. Antigen detection was performed with a tyramide signal amplification-biotin system (Perkin-Elmer, Boston, MA) by following the manufacture's instructions. Chromogenic detection of horseradish peroxidase was performed with a Metal Enhanced 3,3′-diaminobenzidine substrate kit (Pierce Biotechnology, Rockford, IL). Antibodies for MMP2, MMP9, and MT1-MMP (Daiichi Fine Chemical Co., Ltd., Toyama, Japan) and a secondary antibody for mouse (Santa Cruz Biotechnology, Inc., Santa Cruz CA) were purchased commercially. The immunohistochemical samples were counterstained with hematoxylin.
MMP inhibitor and MAPK inhibitor.
MMP2 inhibitor, MMP9 inhibitor, and inhibitors GM6001, PD98059, SP600125, and SB203580 were purchased from Merck KGaA (Darmstadt, Germany). Concentrations were as follows: dimethyl sulfoxide, ×500; MMP2 inhibitor, 20 μM; MMP9 inhibitor, 2 μM; GM6001, 5 μM; PD98059, 20 μM; SP600125, 20 μM; and SB203580, 5 μM. Inhibitors were introduced into the medium mixture for the organotypic raft culture from day 1 to day 10.
RESULTS
E7-expressing keratinocytes escaped from Ras-induced senescence through pRb degradation.
We analyzed the effects of Ras activation on HPV-induced tumorigenesis. First, we introduced the active form of H-ras, which has one amino acid substitution (H-rasV12, consisting of a G-to-V change at position 12), into primary HFKs and analyzed the growth potential (Fig. 1). It was reported that the Ras activation in human primary cells induced premature senescence (49). In agreement with that report, the Ras activation could induce senescence-like growth arrest in HFKs. The Ras activation attenuated the cell growth and changed the cell to a flattened and enlarged senescence-like shape. The senescence induction could also be confirmed by SA-β-gal assay (Fig. 1A). Next, the H-rasV12 was introduced into the HFKs expressing HPV oncoprotein. Although Ras activation could induce senescence-like growth arrest in the HFKs expressing HPV18 E6 (18E6), 18E7 expression rescued the cells from senescence.
FIG. 1.
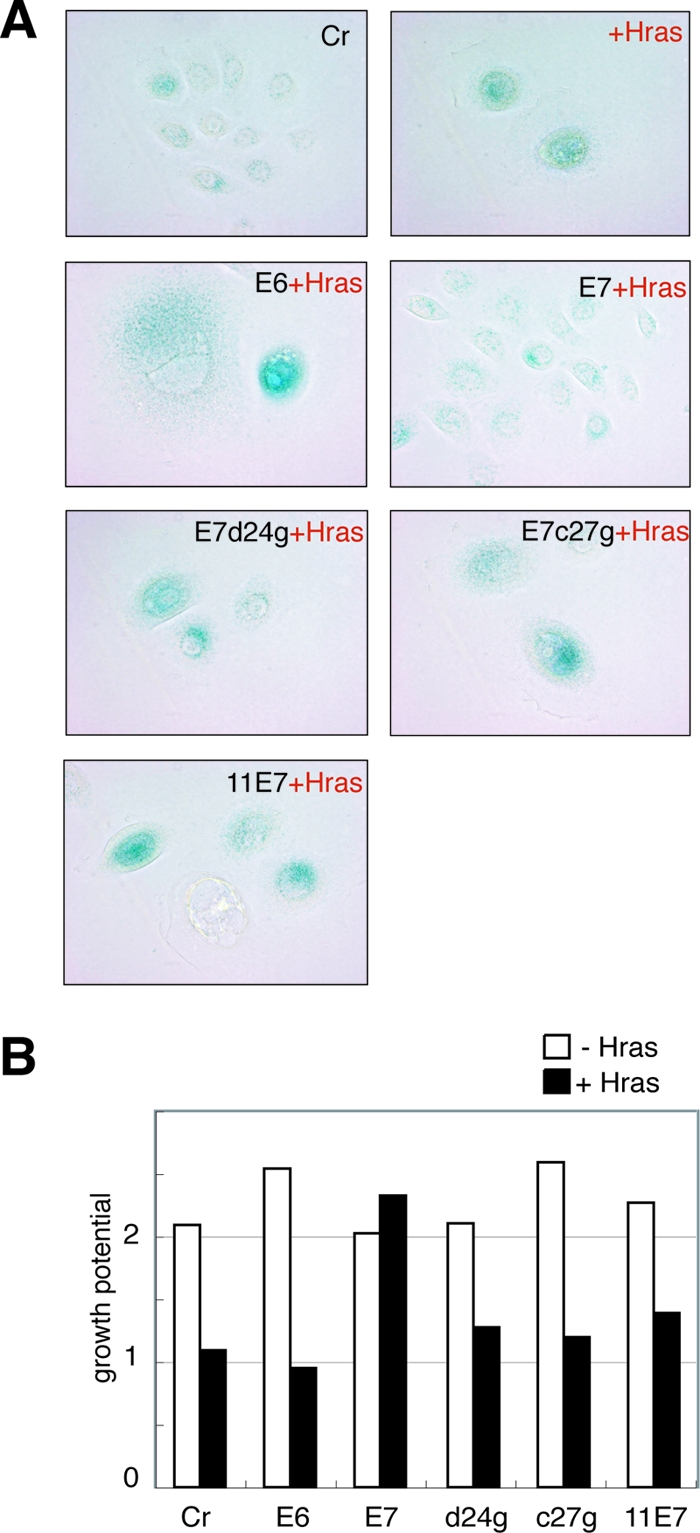
Coexpression of HPV oncoproteins and H-rasV12 in primary HFKs. (A) Morphology and senescence induction of the HFKs expressing HPV oncoproteins and H-ras. The senescent cells were detected by SA-β-gal assay. The control cells (Cr) used in this and the following experiments were infected with two empty retroviral vectors, pLXSN and pLPCX. Magnification, ×200. (B) The effects of H-ras activation on the proliferation of the HFKs expressing HPV oncoproteins. At days 1 and 3 after they were seeded, the cells in the dishes were counted, and relative cell numbers (those at 1 day after seeding are set at 1) are indicated as a bar.
High-risk type E7 binds to and inactivates pRb and induces its degradation through the proteasome pathway (18). To investigate whether pRb inactivation was required for escape from Ras-induced senescence, we analyzed the effects of two 18E7 mutants (D24G and C27G) and low-risk HPV11 E7 (11E7) expression on Ras-induced senescence (Fig. 1). These E7 mutants could not inactivate the pRb (22, 31), and neither of them could eliminate the Ras-induced senescence, indicating that pRb inactivation was important for escape from senescence.
E7 has been reported to bind to other pocket protein family members (37). Although the degradation of pRb is specific to high-risk E7, we reported that p130 was targeted for degradation by both types of E7 (HPV18 E7 and HPV11 E7) in HFKs (52). In the same report, we demonstrated that the expression level of p107 was not modified by E7s. Considering these observations, the results shown above indicate that p107 and p130 were not involved in the Ras-induced senescence. This notion was supported by the observation that a pRb knockdown with a specific shRNA could eliminate the Ras-induced senescence (data not shown).
Coexpression of 18E7 and H-rasV12 conferred invasive potential on epidermis raft culture.
We analyzed the effect of Ras activation on epidermis formation, using an organotypic raft culture system (Fig. 2A). With control HFKs, normal epidermal structure could be reproduced. The cells expressing H-rasV12 or both H-rasV12 and 18E6 could not be used for the construction of the raft culture because their growth potentials were severely retarded, as shown in Fig. 1B. The 18E7 expression induced hyperplasia in the epidermal layer as previously reported (52). The cells expressing both H-rasV12 and 18E7 showed hyperplasia and invaded the dermal layer that consisted of type I collagen and HFFs, suggesting that Ras activation conferred invasive potential on the cells expressing high-risk E7. Invasion induced by H-rasV12 and 18E7 was confirmed by an invasion assay using a Transwell coated with extracellular matrix (Matrigel; BD Biosciences, Franklin Lakes, NJ).
FIG. 2.
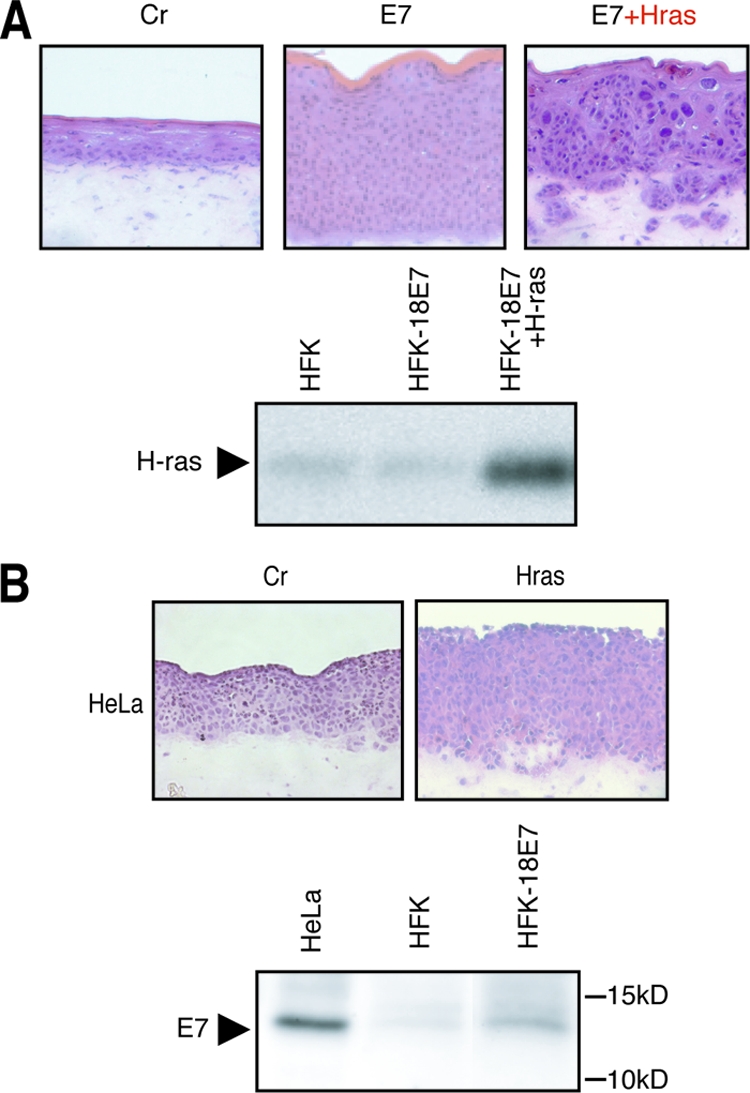
Effects of Ras activation in E7-expressing cells were evaluated using raft culture. (A) HFKs expressing 18E7 and/or H-rasV12 were used for the raft culture construction. The cryosection (7 μm) was fixed with formaldehyde and stained with H&E. Magnifications in this and subsequent figures, ×100. H-ras expression in the cells was monitored by immunoblot analysis. (B) HeLa cells were used for the raft culture system. H-rasV12 was transduced by the retroviral vector pLPCX. 18E7 expression level was analyzed with an 18E7-specific polyclonal antibody. Cr, control.
In contrast to the invasive potential observed for the HFKs, HeLa cells derived from HPV18-positive cervical carcinoma showed very weak invasion activity, even with the ectopic expression of H-rasV12 (Fig. 2B), which raised the possibility that the HFKs expressed E7 protein more abundantly than the HeLa cells. As shown in Fig. 2B, the expressions level of 18E7 in the HFKs was apparently lower than that in HeLa cells, suggesting that the 18E7 expression level in the HFKs was within the physiological level.
Ras activation was reported to contribute to the invasive potential of cancer cells by inducing MMPs through MAPK pathway activation (10). To investigate whether MMPs were induced in the epidermal layer of the raft culture expressing both H-rasV12 and 18E7, we performed immunohistochemical analyses for MMP2 and MMP9, which are type IV collagenases and major MMPs inducible by Ras (Fig. 3). MMP2 was constitutively expressed by the HFFs embedded in the dermal equivalent and was broadly distributed in the dermal layer. The expression of MMP9 was induced in the whole epidermal layer by 18E7 expression. These MMPs might contribute to the invasive potential of the HFKs expressing both H-rasV12 and 18E7.
FIG. 3.

Immunohistochemical analysis of MMP expression in the raft culture. MMP expression was detected by a TSA biotin system and is shown as a dark, brownish signal. The samples were counterstained with hematoxylin. Cr, control.
In the acquisition of invasive potential, the activation of MMPs through cleavage by other proteases is also involved (13). MT1-MMP is a matrix-anchored type of MMP, which is a major activator for MMP2 (26). MT1-MMP can also cleave other MMPs and extracellular matrix proteins, for example, fibronectin, laminin, and collagen, and contributes to cell invasiveness. We analyzed the MT1-MMP expression in the raft culture expressing H-rasV12 and 18E7 (Fig. 3). The expression of MT1-MMP was induced by H-rasV12 in the epidermal keratinocytes, especially at the boundary between the epidermal and the dermal layers. In the same sample, MMP2 also accumulated at the boundary (Fig. 3, E7+Hras panels), suggesting that the MT1-MMP induced by the Ras activation recruited the MMP2 expressed by the HFFs for its activation.
MMP upregulation confers invasive potential on the H-rasV12 and 18E7-coexpressing epidermis.
To investigate whether the MMP induction described above was involved in the invasion potential, we analyzed the effects of MMP inhibitors on invasion (Fig. 4). The MMP2-specific inhibitor partially inhibited invasion; the MMP9-specific inhibitor strongly inhibited invasion, suggesting that the MMP9 induced by 18E7 contributed to invasion. However, induction of MMP9 might not be sufficient for invasion because the expression of 18E7 only could not confer the invasion potential on the keratinocytes. GM6001, which is a broad inhibitor of MMPs, suppressed invasion of MMP9. These results suggested that these MMPs cooperated with each other to promote the invasiveness of the HFKs expressing both H-rasV12 and 18E7.
FIG. 4.

Effects of MMP-specific inhibitors on the invasion induced by both 18E7 and H-rasV12. MMP-specific inhibitors were added to the raft culture medium from day 1 (described in Materials and Methods). The cryosection (7 μm) was fixed with formaldehyde and stained with H&E. Cr, control.
MMP9 and MT1-MMP were induced through the MEK/ERK pathway.
It is known that Ras induces MMP expression through the activation of the MAPK pathway (30). To investigate the involvement of MAPK activation in invasion, the effects of the MAPK-specific inhibitors on invasion potential were examined (Fig. 5). A MEK-specific inhibitor, PD98059, clearly suppressed the invasion capacity of the cells expressing both E7 and H-ras. A p38-specific inhibitor, SB203580, had no effect on invasiveness. SP600125, a JNK-specific inhibitor, inhibited normal epidermis formation, suggesting that the JNK activity was essential for the development of stratified epidermis as previously reported (58). These results indicate that the invasion potential was induced through the activation of the MEK/ERK pathway.
FIG. 5.

Effects of MAPK-specific inhibitors on the invasion induced by both 18E7 and H-rasV12. MAPK-specific inhibitors were added to the raft culture medium from day 1. The cryosection (7 μm) was fixed with formaldehyde and stained with H&E. Cr, control.
Next, the effects of the MAPK inhibitors on the expressions of MMP9 and MT1-MMP were analyzed by immunohistochemical assay (Fig. 6). Expression of MMP9 (Fig. 6A) and MT1-MMP (Fig. 6B) was suppressed by PD98059, suggesting that their expression was induced through the activation of the MEK/ERK pathway.
FIG. 6.

Effects of the MAPK-specific inhibitors on the MMP expression. The MAPK-specific inhibitors were added to the raft culture medium from day 1. Immunohistochemical analyses were performed for MMP9 (A) and MT1-MMP (B). The samples were counterstained with hematoxylin. Cr, control.
18E7 and constitutive active MEK1-coexpressing epidermis partly reproduced the invasiveness of the H-rasV12 and HPV18 E7-coexpressing epidermis.
The results shown above suggested that the invasiveness induced by H-rasV12 and 18E7 was mediated by the MMP activation through the MEK/ERK pathway. To examine whether the MEK/ERK activation could mimic the Ras activation for the induction of invasiveness, the constitutively active mutant of MEK1, MKK7 or MKK6, was expressed in the HFKs expressing 18E7, and the raft culture was constructed with them (Fig. 7). The epidermis expressing MEK1-SDSE (the MEK1 constitutive active mutant) acquired invasive potential, and MT1-MMP expression was induced. MKK7-DED (for the JNK pathway) and MKK6-SETE (for the p38 pathway) expression did not induce the invasion. Although these results indicated the involvement of the MEK/ERK pathway in the induction of MT1-MMP and invasiveness, the invasion induced by the MEK1-SDSE was attenuated compared with that induced by H-rasV12 and 18E7 coexpression, suggesting that other pathways or factors in addition to MAPK activation contribute to the invasive potential in Ras activation. It was apparent that all of the MAPK mutants expressing epidermis showed a thickened cornified layer, suggesting that these MAPK pathways contribute to the differentiation program of epidermis in some part.
FIG. 7.

Expressions of constitutive active MAPKs in the HPV18 E7-expressing keratinocytes. Invasion activity was monitored with the organotypic raft culture. The cryosection (7um) was fixed with formaldehyde and stained with H&E. MMP expression was monitored by immunohistochemical analysis, and the samples were counterstained with hematoxylin (MMP9 and MT1-MMP).
MMP9 induction was independent of pRb degradation.
We investigated the mechanisms by which 18E7 induced MMP9 expression. A well-known activity of the high-risk E7 is its capacity to bind to and inactivate pRb. We therefore examined the effect of the pRb knockdown on the expression of MMP9. The raft culture with the pRb knockdown cells was established by using a retroviral vector expressing the shRNA targeted to pRb. The pRb expression level was monitored by immunoblot analysis with anti-pRb antibody (Fig. 8A). The pRb knockdown did not induce MMP9 in the epidermis, and the expression of low-risk HPV11 E7 that could not inactivate pRb induced MMP9 (Fig. 8B), suggesting that the upregulation of MMP9 by E7 was independent of the pRb degradation.
FIG. 8.
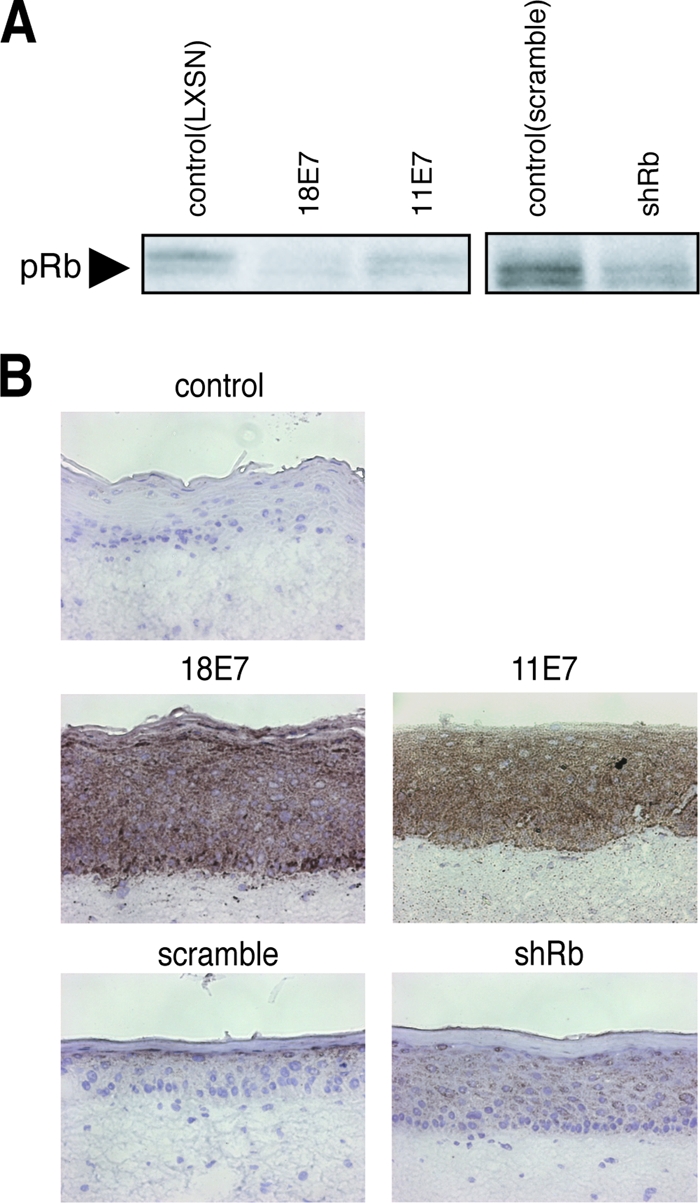
Involvement of pRb downregulation in the E7-induced MMP9 expression. (A) The effect on pRb expression by E7 (derived from pLXSN) or shRNA targeted to pRb (shRb) (derived from pSIREN-RetroQ) was analyzed by immunoblotting. (B) HFKs expressing E7 or shRb were used to construct the raft culture. A random oligonucleotide was used as a control for the shRb (scramble).
DISCUSSION
Cancer-prone status of HPV-infected cells.
For HPV-induced carcinogenesis, the major viral oncoproteins E6 and E7 are pivotal; both E6 and E7 disturb the cell cycle checkpoint and the apoptosis induction, resulting in the augmentation of genetic instability. For the progression of carcinogenesis, it is believed that additional host mutations are required and that the ras family of proto-oncogenes is one of the major targets for the mutations. Although Ras activation in HFKs induces premature senescence through the pRb-related pathway, the high-risk E7 oncoprotein could eliminate senescence induction and confer invasive potential on the HFKs in cooperation with Ras activation (Fig. 1 and 2). The results indicate that the HPV-infected cells that express E7 are tolerant of the Ras activation, which contributes to malignant conversion of the HPV-infected lesion. Other oncogenic stresses, such as the activations of Raf, ERK, or Akt, are also reported to induce “premature senescence” (5), and our observation suggested that the high-risk HPV-infected cells are tolerant of such stresses, which might confer the “cancer-prone” status on the HPV-infected cells.
It was reported that transformation by Ras activation in normal human cells requires p53 inactivation, pRb inactivation, c-myc activation, PP2A inactivation, and telomerase reactivation (20). Although our report indicated that the high-risk E7 expression, which induces pRb degradation and c-myc activation, conferred invasiveness on the HFKs in cooperation with the Ras activation, the combined expression of 18E7 and H-rasV12 is not sufficient for the induction of transformation. For the transformation, additional alterations of the host cells as well as high-risk E6 expression must be required. We are seeking such additional cellular factors that are involved in the multiple aspects of the malignant conversion, such as the cell motility, the metastasis conversion, and the evasion of the host immune system.
MMP induction and invasiveness.
The acquisition of invasive potential by the coexpression of H-rasV12 and 18E7 resulted from MMP induction, which was supported by experiments with several MMP inhibitors (Fig. 4). MMP9 expression was upregulated by 18E7, but 18E7 expression by itself did not induce the invasiveness of the epidermal cells (Fig. 2, 3). Although this observation indicated that MMP9 expression was not sufficient for the acquisition of the invasive potential, its involvement in invasiveness was supported by the result of the MMP9-specific inhibitor (Fig. 4). It was reported that MMP9 was related not only to invasiveness but also to cell proliferation (8). The hyperplasia of epidermis induced by 18E7 expression was significantly suppressed by the MMP9-specific inhibitor (Fig. 4), suggesting that the MMP9 expression was also involved in the cell proliferation of epidermis. We reported that the 18E7-induced hyperplasia resulted from the degradation of pRb and p130 and that the low-risk HPV11 E7 caused mild hyperplasia by inactivating p130 (52). The induction of MMP9 was independent of pRb inactivation, and HPV11 E7 had a potential to induce MMP9 expression (Fig. 8), indicating a possibility that MMP9 induction was a common feature of both types of E7 and that induction is involved in E7-mediated hyperplasia.
E7-mediated MMP induction has been reported with other types of HPVs. HPV16 E7 induced MT1-MMP expression in keratinocytes; such an effect was not evident for HPV18-positive HeLa cells (50, 57). It was reported that the expression of HPV8 E7, which is a cutaneous-type HPV, was enough to confer the invasive potential on the epithelial cells in the organotypic raft culture (1), in which MMP1, MMP8, and MT1-MMP were induced. The differences between E7 functions in the invasiveness of various HPV types might be due to their host cell tropism, and it will be necessary to examine the key factor involved in the MMP induction by E7s. An examination of the expression levels of a variety of MMPs in the epidermal cells expressing both 18E7 and H-ras will also be required.
MT1-MMP was induced by H-rasV12 in the epidermal cells of the raft culture, especially at the basal layer cells in contact with the dermal equivalent (Fig. 3). It was reported that MT1-MMP was highly expressed at the cell surface of cancer cells (26). MMP2 was expressed from the fibroblasts embedded in the dermal equivalent and was broadly distributed in the dermal layer (Fig. 3), which is one of the proteolytic targets of MT1-MMP. The immunohistochemical staining showed the accumulation of MMP2 at the region of basal membrane, suggesting that the MT1-MMP recruited the MMP2 at the region and activated it by proteolytic cleavage (51). It is known that MT1-MMP is also able to degrade type I collagen (24). Type I collagen is a major component of the dermal region (6); therefore, the MT1-MMP might function as a “pioneer” for the invasion into the dermal region.
In contrast to MT1-MMP, both MMP2 and MMP9 are known as type IV collagenases (13, 53). Type IV collagen localizes at the basal membrane (6), and both MMPs might be involved in the degradation of basal membrane at the early stage of invasion. MMP2 was also known to activate MMP9 (16). A recent study reported that MMP2 could upregulate cell migration in vitro in cooperation with MMP9 (42). That report, using the knockout mouse for MMP2 and MMP9, indicated their contribution to the invasiveness (33). These reports indicate that a cooperative function between MMP2 and MMP9 is important for invasiveness.
The MEK/ERK pathway was involved in the MMP induction by H-ras and 18E7.
The expression of MMP9 and MT1-MMP was upregulated through the MEK/ERK pathway. The upstream events, however, might be distinct, because they were differentially regulated by 18E7 and H-ras (Fig. 3). The constitutive active mutant of MEK could induce significant but weak invasion in cooperation with 18E7 (Fig. 7), suggesting that the other pathways triggered by the Ras activation were involved in the strong invasiveness observed for the cells expressing both H-rasV12 and 18E7. The p38 pathway appeared not to be involved in the epidermal formation and the invasion potential (Fig. 5). JNK activation seemed to be essential for the proliferation of stratified epithelial cells, because SP600125, a JNK inhibitor, drastically suppressed epidermal formation, which is consistent with a report that the JNK activity was essential for the development of stratified epidermis (17, 58).
We reproduced a portion of multistep carcinogenesis, “the acquisition of invasiveness,” by introducing 18E7 and H-rasV12 into the raft culture system. The raft culture is suitable to analyze the cell invasion and motility in a semiphysiological condition and useful for the screening of the drugs against invasiveness.
Acknowledgments
This work is supported by Japanese Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology and by the Japanese Ministry of Health and Welfare. S.Y. and H.N. are supported by the 21st Century COE Program of JSPS.
Activated H-ras gene was provided by Ishikawa. Several DNAs of MAPK mutants were provided by Nishida.
Footnotes
Published ahead of print on 25 June 2008.
REFERENCES
- 1.Akgül, B., R. García-Escudero, L. Ghali, H. J. Pfister, P. G. Fuchs, H. Navsaria, and A. Storey. 2005. The E7 protein of cutaneous human papillomavirus type 8 causes invasion of human keratinocytes into the dermis in organotypic cultures of skin. Cancer Res. 652216-2223. [DOI] [PubMed] [Google Scholar]
- 2.Alonio, L. V., M. A. Dalbert, D. Mural, J. Bartt, O. Bazán, G. Dominguez, and A. R. Teyssié. 2003. Ha-ras oncogene mutation associated to progression of papillomavirus induced lesions of uterine cervix. J. Clin. Virol. 27263-269. [DOI] [PubMed] [Google Scholar]
- 3.Bos, J. L. 1989. ras oncogenes in human cancer: a review. Cancer Res. 494682-4689. [PubMed] [Google Scholar]
- 4.Cairns, J. 1975. Mutation selection and the natural history of cancer. Nature 255197-200. [DOI] [PubMed] [Google Scholar]
- 5.Campisi, J., and F. d'Adda di Fagagna. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8729-740. [DOI] [PubMed] [Google Scholar]
- 6.Canty, E. G., and K. E. Kadler. 2005. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 1181341-1353. [DOI] [PubMed] [Google Scholar]
- 7.Cole, S. T., and O. Danos. 1987. Nucleotide sequence and comparative analysis of the human papillomavirus type 18 genome. Phylogeny of papillomaviruses and repeated structure of the E6 and E7 gene products. J. Mol. Biol. 193599-608. [DOI] [PubMed] [Google Scholar]
- 8.Coussens, L. M., C. L. Tinkle, D. Hanahan, and Z. Werb. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103481-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dollard, S. C., J. L. Wilson, L. M. Demeter, W. Bonnez, R. C. Reichman, T. R. Broker, and L. T. Chow. 1992. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. Genes Dev. 61131-1142. [DOI] [PubMed] [Google Scholar]
- 10.Downward, J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 311-22. [DOI] [PubMed] [Google Scholar]
- 11.Duensing, S., A. Duensing, E. R. Flores, A. Do, P. F. Lambert, and K. Münger. 2001. Centrosome abnormalities and genomic instability by episomal expression of human papillomavirus type 16 in raft cultures of human keratinocytes. J. Virol. 757712-7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyson, N., P. M. Howley, K. Münger, and E. Harlow. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243934-937. [DOI] [PubMed] [Google Scholar]
- 13.Egeblad, M., and Z. Werb. 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2161-174. [DOI] [PubMed] [Google Scholar]
- 14.Fehrmann, F., and L. A. Laimins. 2003. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene 225201-5207. [DOI] [PubMed] [Google Scholar]
- 15.Flores, E. R., and P. F. Lambert. 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 717167-7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridman, R., M. Toth, D. Pena, and S. Mobashery. 1995. Activation of progelatinase B (MMP-9) by gelatinase A (MMP-2). Cancer Res. 552548-2555. [PubMed] [Google Scholar]
- 17.Gazel, A., T. Banno, R. Walsh, and M. Blumenberg. 2006. Inhibition of JNK promotes differentiation of epidermal keratinocytes. J. Biol. Chem. 28120530-20541. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez, S. L., M. Stremlau, X. He, J. R. Basile, and K. Münger. 2001. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 757583-7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenhalgh, D. A., X. J. Wang, J. A. Rothnagel, J. N. Eckhardt, M. I. Quintanilla, J. L. Barber, D. S. Bundman, M. A. Longley, R. Schlegel, and D. R. Roop. 1994. Transgenic mice expressing targeted HPV-18 E6 and E7 oncogenes in the epidermis develop verrucous lesions and spontaneous, Ha-ras-activated papillomas. Cell Growth Differ. 5667-675. [PubMed] [Google Scholar]
- 20.Hahn, W. C., C. M. Counter, A. S. Lundberg, R. L. Beijersbergen, M. W. Brooks, and R. A. Weinberg. 1999. Creation of human tumour cells with defined genetic elements. Nature 400464-468. [DOI] [PubMed] [Google Scholar]
- 21.Harada, T., O. Matsuzaki, H. Hayashi, S. Sugano, A. Matsuda, and E. Nishida. 2003. AKRL1 and AKRL2 activate the JNK pathway. Genes Cells 8493-500. [DOI] [PubMed] [Google Scholar]
- 22.Heck, D. V., C. L. Yee, P. M. Howley, and K. Münger. 1992. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 894442-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helt, A. M., J. O. Funk, and D. A. Galloway. 2002. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J. Virol. 7610559-10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hotary, K., E. Allen, A. Punturieri, I. Yana, and S. J. Weiss. 2000. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 1491309-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howley, P. M., and D. Lowy. 2001. Papillomaviruses and their replication, p. 2197-2229. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Baltimore, MD.
- 26.Itoh, Y., and M. Seiki. 2006. MT1-MMP: a potent modifier of pericellular microenvironment. J. Cell Physiol. 2061-8. [DOI] [PubMed] [Google Scholar]
- 27.Jones, D. L., R. M. Alani, and K. Münger. 1997. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 112101-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiyono, T., A. Hiraiwa, M. Fujita, Y. Hayashi, T. Akiyama, and M. Ishibashi. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 9411612-11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klingelhutz, A. J., S. A. Foster, and J. K. McDougall. 1996. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 38079-82. [DOI] [PubMed] [Google Scholar]
- 30.Kranenburg, O., M. F. Gebbink, and E. E. Voest. 2004. Stimulation of angiogenesis by Ras proteins. Biochim. Biophys. Acta 165423-37. [DOI] [PubMed] [Google Scholar]
- 31.Lee, J. O., A. A. Russo, and N. P. Pavletich. 1998. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391859-865. [DOI] [PubMed] [Google Scholar]
- 32.Lee, S. S., R. S. Weiss, and R. T. Javier. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 946670-6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masson, V., L. R. de la Ballina, C. Munaut, B. Wielockx, M. Jost, C. Maillard, S. Blacher, K. Bajou, T. Itoh, S. Itohara, Z. Werb, C. Libert, J. M. Foidart, and A. Noël. 2005. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 19234-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medína-Martinez, O., V. Vallejo, M. C. Guido, and A. García-Carrancá. 1997. Ha-ras oncogene-induced transcription of human papillomavirus type 18 E6 and E7 oncogenes. Mol. Carcinog. 1983-90. [PubMed] [Google Scholar]
- 35.Meyers, C., M. G. Frattini, J. B. Hudson, and L. A. Laimins. 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257971-973. [DOI] [PubMed] [Google Scholar]
- 36.Münger, K., A. Baldwin, K. M. Edwards, H. Hayakawa, C. L. Nguyen, M. Owens, M. Grace, and K. Huh. 2004. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 7811451-11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Münger, K., J. R. Basile, S. Duensing, A. Eichten, S. L. Gonzalez, M. Grace, and L. V. Zancy. 2001. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 207888-7898. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen, M. L., M. M. Nguyen, D. Lee, A. E. Griep, and P. F. Lambert. 2003. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J. Virol. 776957-6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Connor, M. J., W. Stünkel, C. H. Koh, H. Zimmermann, and H. U. Bernard. 2000. The differentiation-specific factor CDP/Cut represses transcription and replication of human papillomaviruses through a conserved silencing element. J. Virol. 74401-410. [PMC free article] [PubMed] [Google Scholar]
- 40.Pietenpol, J. A., R. W. Stein, E. Moran, P. Yaciuk, R. Schlegel, R. M. Lyons, M. R. Pittelkow, K. Münger, P. M. Howley, and H. L. Moses. 1990. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 61777-785. [DOI] [PubMed] [Google Scholar]
- 41.Pim, D., and L. Banks. 1991. Loss of HPV-16 E7 dependence in cells transformed by HPV-16 E7 plus EJ-ras correlates with increased c-myc expression. Oncogene 6589-594. [PubMed] [Google Scholar]
- 42.Robinson, C. M., A. M. Stone, J. D. Shields, S. Huntley, I. C. Paterson, and S. S. Prime. 2003. Functional significance of MMP-2 and MMP-9 expression by human malignant oral keratinocyte cell lines. Arch. Oral Biol. 48779-786. [DOI] [PubMed] [Google Scholar]
- 43.Ruesch, M. N., F. Stubenrauch, and L. A. Laimins. 1998. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J. Virol. 725016-5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sagae, S., R. Kudo, N. Kuzumaki, T. Hisada, Y. Mugikura, T. Nihei, T. Takeda, and M. Hashimoto. 1990. Ras oncogene expression and progression in intraepithelial neoplasia of the uterine cervix. Cancer 66295-301. [DOI] [PubMed] [Google Scholar]
- 45.Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M. Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 631129-1136. [DOI] [PubMed] [Google Scholar]
- 46.Schiffman, M., P. E. Castle, J. Jeronimo, A. C. Rodriguez, and S. Wacholder. 2007. Human papillomavirus and cervical cancer. Lancet 370890-907. [DOI] [PubMed] [Google Scholar]
- 47.Schreiber, K., R. E. Cannon, T. Karrison, G. Beck-Engeser, D. Huo, R. W. Tennant, H. Jensen, W. M. Kast, T. Krausz, S. C. Meredith, L. Chen, and H. Schreiber. 2004. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene 233972-3979. [DOI] [PubMed] [Google Scholar]
- 48.Schubbert, S., K. Shannon, and G. Bollag. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7295-308. [DOI] [PubMed] [Google Scholar]
- 49.Serrano, M., A. W. Lin, M. E. McCurrach, D. Beach, and S. W. Lowe. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88593-602. [DOI] [PubMed] [Google Scholar]
- 50.Smola-Hess, S., J. Pahne, C. Mauch, P. Zigrino, H. Smola, and H. J. Pfister. 2005. Expression of membrane type 1 matrix metalloproteinase in papillomavirus-positive cells: role of the human papillomavirus (HPV) 16 and HPV8 E7 gene products. J. Gen. Virol. 861291-1296. [DOI] [PubMed] [Google Scholar]
- 51.Taniwaki, K., H. Fukamachi, K. Komori, Y. Ohtake, T. Nonaka, T. Sakamoto, T. Shiomi, Y. Okada, T. Itoh, S. Itohara, M. Seiki, and I. Yana. 2007. Stroma-derived matrix metalloproteinase (MMP)-2 promotes membrane type 1-MMP-dependent tumor growth in mice. Cancer Res. 674311-4319. [DOI] [PubMed] [Google Scholar]
- 52.Ueno, T., K. Sasaki, S. Yoshida, N. Kajitani, A. Satsuka, H. Nakamura, and H. Sakai. 2006. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 254155-4164. [DOI] [PubMed] [Google Scholar]
- 53.Werb, Z. 1997. ECM and cell surface proteolysis: regulating cellular ecology. Cell 91439-442. [DOI] [PubMed] [Google Scholar]
- 54.Woodman, C. B., S. I. Collins, and L. S. Young. 2007. The natural history of cervical HPV infection: unresolved issues. Nat. Rev. Cancer 711-22. [DOI] [PubMed] [Google Scholar]
- 55.Yokota, J., and T. Sugimura. 1993. Multiple steps in carcinogenesis involving alterations of multiple tumor suppressor genes. FASEB J. 7920-925. [DOI] [PubMed] [Google Scholar]
- 56.Yukawa, K., K. Butz, T. Yasui, H. Kikutani, and F. Hoppe-Seyler. 1996. Regulation of human papillomavirus transcription by the differentiation-dependent epithelial factor Epoc-1/skn-1a. J. Virol. 7010-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhai, Y., K. B. Hotary, B. Nan, F. X. Bosch, N. Muñoz, S. J. Weiss, and K. R. Cho. 2005. Expression of membrane type 1 matrix metalloproteinase is associated with cervical carcinoma progression and invasion. Cancer Res. 656543-6550. [DOI] [PubMed] [Google Scholar]
- 58.Zhang, J. Y., C. L. Green, S. Tao, and P. A. Khavari. 2004. NF-κB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 117-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.zur Hausen, H. 1996. Papillomavirus infections: a major cause of human cancers. Biochim. Biophys. Acta 1288F55-F78. [DOI] [PubMed] [Google Scholar]