

Abstract
An efficient and general procedure was developed for the direct alkylation of H-phosphinate esters with LiHMDS at low temperature. The simplicity of the reaction allows the use of various H-phosphinate esters and takes place with a wide range of electrophiles. The approach can be employed to access some GABA analogs or precursors to GABA analogs. The isolated yields are moderate to good. This is the first report of an alkylation with a secondary iodide, or a primary chloride.
Keywords: Alkylation, H-Phosphinate, Phosphinic Acid, Lithium, Hexamethyldisilazide
Introduction
Over the years, many examples of base-promoted H-phosphinate alkylation (eq 1) have been reported in the literature.1 However, there does not appear to be a standard set of conditions, and surprisingly, we have not found any general study of this reaction. Various bases (RONa, NaH, BuLi, LDA, KHMDS) and stoichiometries have been employed.1 A somewhat more widely employed approach (eq 2) consists of silylating H-phosphinic acids, followed by an Arbuzov-like reaction with alkyl halides.2 This method also has its limitations, and it often requires esterification of the dialkylphosphinic acid products when further manipulations are desired.
(1).
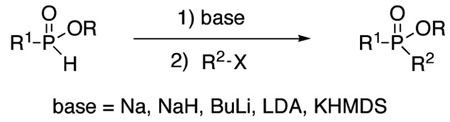
(2).

Functionalized, differentially-substituted phosphinate esters R1R2P(O)(OR) are important organophosphorus intermediates, particularly in the synthesis of medicinally-relevant protease inhibitors and ATP-dependent ligases.3 Over the past few years, our laboratory has been developing various approaches to prepare H-phosphinic acids and esters.4 With these compounds becoming more widely available, we are turning our focus to the functionalization of these intermediates with the formation of a second phosphorus-carbon bond. Our literature survey uncovered the lack of general conditions for the base-promoted alkylation of H-phosphinates with alkyl halides. We therefore decided to investigate the scope and limitations of this transformation. Herein, we report a detailed investigation which led to a standardized set of conditions allowing the preparation of functionalized dialkylphosphinates.
Results and Discussion
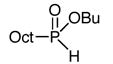
Butyl octyl-H-phosphinate was selected as test substrate to determine the choice of base, with n-butyl iodide as the electrophile (Table 1). The phosphorus nucleophile, butyl iodide, and base were used in equimolar quantities, and the results studied by 31P-NMR of the crude reaction mixtures. Although alkylation takes place in all cases, significant differences are observed. Not surprisingly, nucleophilic bases, such as MeLi and BuLi (entries 3 and 4), give lower yields due to the competing direct substitution of the butyl ester to form a secondary phosphine oxide, whereas the strong non-nucleophilic bases (entries 5 and 6) give better results. After this initial screening, LiHMDS was selected to investigate the influence of the electrophile.
Table 1.
a Role of the base in the alkylation of butyl octyl-H-phosphinate with butyl iodide
| Entry | Substrate | Conditions | Base | NMR conversion%b |
|---|---|---|---|---|
| 1 | BuI | THF, 0 °C to rt | Na | 16 |
| 2 | BuI | THF, −78 °C to rt | i-PrMgCl | 64 |
| 3 | BuI | THF, −78 °C to rt | MeLi | 56 |
| 4 | BuI | THF, −78 °C to rt | BuLi | 60 |
| 5 | BuI | THF, −78 °C to rt | LDA | 89 |
| 6 | BuI | THF, −78 °C to rt | LiHMDS | 100 |
All reactions were conducted in freshly distilled anhydrous THF, under N2.
NMR conversion yields are determined by integration of all the resonances in the crude 31P NMR spectra. investigate the influence of the electrophile
Table 2 summarizes the results obtained in the LiHMDS-mediated alkylation of PhP(O)(OEt)H with various alkyl halides. Under otherwise identical conditions, a clear erosion in yield is observed as the reactivity of the electrophile decreases. 31P-NMR analysis of these reaction mixtures indicate the formation of PhP(O)(OEt)OLi (δ17.2 ppm), along with the P(III) anion PhP(OEt)(OLi) (δ 149 ppm), and the alkylation product PhP(O)(OEt)Oct (δ 44.8 ppm). The low reactivity of the electrophile requires heating the reaction mixture during which competitive oxidation of the anion takes place, so a deoxygenation protocol was investigated.5 Instead of a rigorous freeze-thaw-degass cycle, we opted for a simpler, more practical, deoxygenation method consisting of placing the reaction flask containing the H-phosphinate ester and THF under vacuum for 5 min at −78 °C, and filling it with nitrogen prior to adding the other reagents. This moderate deoxygenation is sufficient to provide good alkylation yields with unreactive electrophiles (X = Cl, OTs). To the best of our knowledge, this is the first example of successful alkylation with an alkyl chloride. Unlike primary iodides which are sufficiently reactive to not necessitate deoxygenation, isopropyl iodide behaves like n-octyl chloride and tosylate. This is also the first time a secondary halide is employed in the alkylation of a H-phosphinate.
Table 2.
Role of the electrophile in the alkylation of PhP(O)(OEt)H with LiHMDS
| Entry | RX | Deoxygenationa | Temperature | NMR conversion%b | Isolated yield %c |
|---|---|---|---|---|---|
| 1 | CH3I | No | −78 °C to rt | 100 | 98 |
| 2 | OctI | No | −78 °C to rt | 100 | 80 |
| 3 | OctBr | No | −78 °C to rt | 80 | 57 |
| 4 | OctBr | Yes | −78 °C to rt | 92 | 71 |
| 5 | OctCl | No | −78 °C to reflux | 1 | - |
| 6 | OctCl | Yes | −78 °C to reflux | 77 | 51 |
| 7 | OctOTs | Yes | −78 °C to reflux | 88 | 62 |
| 8 | i-PrI | Yes | −78 °C to reflux | 87 | 45 |
Deoxygenation was conducted by placing a THF solution of the H-phosphinate under vacuum at −78 C for 5 min, then adding N2.
NMR conversion yields are determined by integration of all the resonances in the 31P NMR spectra.
Isolated yield of pure compounds after chromatography on silica gel.
The next stage in this study was to investigate the scope with respect to both H-phosphinate starting material and electrophile (Table 3).
Table 3.
Reaction Scopea
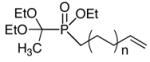
| Entry | H-Phosphinate Ester | Electrophileb | Product | Isolated yield %c |
|---|---|---|---|---|
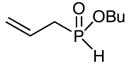
| 1a |

|
|
|
68 |
| 1b | 66 | |||
| 2 |

|
|

|
62 |
| 3 |

|

|

|
70 |
| 4 |

|

|

|
48 |
| 5a |

|
|

|
50 |
| 5b | 70 | |||
| 6a |

|

|

|
48 |
| 6b | 60 | |||
| 6c | 62 | |||
| 7 |

|

|

|
58 |
| 8 |

|

|

|
81 |
| 9 |

|
F2CHCI |
|
71 |
| 10 |

|
F2CHCI |

|
78 |
| 11 |
|
i-Prl |

|
73 |
| 12 |

|
|

|
66 |
| 13 |
|
CH3I |

|
95 |
| 14 |

|
F2CHCL |
|
72 |
| 15a |

|

|

|
50 |
| 15b | 62 | |||
| 15c | 68 |
Details are provided in the experimental section.
Electrophiles which did not react successfully under a variety of conditions (excess of base, heating) include CH2I2, (CH2Br)2, bromoacetates, and (EtO)2P(O)CF2Br. The reasons for failure are unclear.
Isolated yield of pure compounds after chromatography on silica gel.
As shown in Table 3, the general conditions can be successfully applied to a range of H-phosphinate/electrophile pairs. The alkylation of the “Ciba-Geigy reagent” (CH3C(OEt)2P(O)(OEt)H)6 was examined in some detail (entries 1–9), in part because it is a representative H-phosphinate ester, but also because the ketal group can be cleaved to unmask a P(O)H functional group (eq 3).7 While we8 and Gallagher9 have reported the direct alkylation of phosphinates ROP(O)H2 under basic conditions which would provide the H-phosphinate esters in one step (eq 4), the scope of this reaction is limited to reactive electrophiles, such as allylic halides, and it fails with most of the electrophiles used in Table 3.
(3).

(4).

The H-phosphinate starting materials were generally available through our various methodologies (radical, Ni- and Pd- catalyzed hydrophosphinylations, or Pd-catalyzed cross-coupling).4 Various functional groups (esters, imides, carbamates) are tolerated under the reaction conditions (Table 3). This alkylation is comparable or superior to other conditions reported,1 especially because it proceeds with equimolar amounts of reagents. In the literature, an excess of reagent (electrophile or H-phosphinate) is often employed.1 For example, the product in Table 3, entry 2 was obtained in 42% yield, using NaH as the base. With halomethylpyridine hydrohalides (Table 3, entries 6 and 15), an equivalent of LiHMDS is employed prior to adding the electrophile to the lithium phosphinate solution. The products are precursors to GABA analogs, although none showed any significant activity on the GABA-B receptor after appropriate deprotection steps.10
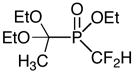
Difluorochloromethane can be used to prepare the corresponding difluoromethylphosphinate. There was an example of such reaction in the literature although the product was not isolated.11 We have extended this to several substrates (Table 3, entries 9, 10, 14). Our results concerning the reactivity of these compounds for the synthesis of fluorinated phosphinates will be reported shortly.
Epoxides also react satisfactorily in the presence of a stoichiometric amount of boron trifluoride etherate (Scheme 1). Products 1 and 2 are obtained as nearly 1:1 mixtures of diastereoisomers because the phosphorus atom is stereogenic. Although epoxide-openings have been reported using the silylation approach, we could not find any report of this reaction under basic conditions.12
Scheme 1.

Epoxide opening
(5).

Equation 5 shows an example of an intramolecular alkylation to form P- heterocycle 3 (1-butoxy-phosphinane-1-oxide).
The based-promoted alkylation can be applied to the synthesis of a variety of biologically active targets. For example, CGP 36742,1c,13 a GABA-B antagonist which is currently undergoing Phase II clinical trials, can be synthesized easily using our radical-based hydrophosphinylation,14 followed by our alkoxysilane-based esterification to form 4,15 and the present alkylation reaction to form intermediate 5. Debenzylation affords CGP 36742 6 cleanly without the need for cumbersome ion-exchange purification.
Another example is shown in Scheme 3 for the rapid synthesis of the known kynureninase inhibitor 8.16 The key step forming 7 proceeds in good yield.
Scheme 3.

Synthesis of a known kynureninase inhibitor
A similar strategy could be applied for the preparation of protected phosphinothricin (Scheme 4).17 The starting H-phosphinate 9 was prepared using our recently disclosed alkylation of phosphinate esters.8 The base-promoted alkylation of 9 delivered protected phosphinothricin 10 in moderate yield.
Scheme 4.

Synthesis of a phosphinothricin precursor
Conclusion
Our investigations of the base-promoted alkylation of H-phosphinate esters reveal that a standardized set of conditions can be established, using LiHMDS as the base and stoichiometric amounts of the reagents. A moderate, yet practical, deoxygenation protocol is necessary with less reactive electrophiles. A wide variety of H-phosphinate esters can be alkylated, including the first successful examples with a primary alkyl chloride and a secondary alkyl iodide electrophiles. The reaction provides a viable alternative to the Arbuzov-like silylation methodology, and it can be applied to the synthesis of functionalized disubstituted phosphinate esters. The method should be useful to phosphorus chemistry practitioners, particularly because the stoichiometry allows the use of valuable moieties, present both in the phosphorus nucleophile and the carbon electrophile.
Experimental Section
Typical Alkylation Procedure
Neat alkyl H-phosphinate ester (1.0 eq., 1.5 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. 5 ml of anhydrous THF was then added under nitrogen. The flask was then placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 1.0 eq., 1.5 mmol) was added at −78°C. After 15 min, the electrophile* (1 eq, 1.5 mmol) was added under N2 as a neat liquid, or as a 0.5 M THF solution for solids. After the addition of the electrophile, the temperature of the solution was slowly allowed to warm to rt.** Once at rt, the reaction mixture was quenched with a satured solution of NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography over silica gel.
For alkyl iodides and alkyl triflate: 10 min at rt (except for the hindered iodides: reflux, 6 h)
For alkyl bromides: 2 to 4 h at rt
For alkyl chlorides, and tosylates : reflux, 6 h, or rt overnight (except for BOMCl : 10 min at rt).
For HCF2Cl the reaction mixture is warmed up and quenched at 0°C.
General procedure for alkyl chlorides and tosylates (Table 2, entry 6). Octyl-phenyl-phosphinic acid ethyl ester.18
Neat ethyl phenyl-H-phosphinate (0.510 g, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. After 10 min, n-octyl chloride (510 μL, 3 mmol) was added under N2. After addition, the reaction mixture was slowly allowed to warm up to rt. The solution was then refluxed overnight under N2. After cooling, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc:Hexanes 40:60) to afford the desired product (51%). RN : [119079-17-3] 1H NMR (CDCl3, 300 MHz) δ 0.85 (t, J = 6.7 Hz, 3 H), 1.22–1.35 (m, 10 H), 1.29 (t, J = 7.0 Hz, 3 H), 1.46–1.60 (m, 2 H), 1.81– 2.04 (m, 2 H), 4.84 & 4.08 (m, 2 H), 7.45–7.60 (m, 3 H), 7.76–7.85 (m, 2 H).
General procedure for alkyl bromides (Table 2, entry 4)
Neat ethyl phenyl-H-phosphinate (0.510 g, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. After 10 min, n-octyl bromide (520 μL, 3 mmol) was added under N2. After addition, the temperature of the solution was slowly allowed to warm to rt. After 3h at rt, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc:Hexanes 40:60) to afford octyl-phenyl-phosphinic acid ethyl ester in 71%.
General procedure for alkyl iodides and triflates (Table 3, entry 8 ). [(1,1-Diethoxy-ethyl)-ethoxy-phosphinoylmethyl]-phosphonic acid diethyl ester.19
Neat ethyl (l,l-diethoxyethyl)phosphinate (630 mg, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was backfilled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. After 10 min, alkyl triflate (0.945 g, 3.15 mmol) dissolved in THF (6 mL) was added under N2. After addition, the temperature of the solution was slowly allowed to warm to rt. After 1 hour at rt, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc:MeOH 95:5) to afford the desired product (81%). RN : [179015-83-9]. 1H NMR (CDCl3, 300 MHz) δ 1.20 & 1.21 (2×t, J = 7.0 Hz, 6 H), 1.34 (m, 9 H), 1.53 (d, JHP = 12.6 Hz, 3 H), 2.41 (ddd, JHP = 20.8 Hz, JHP = 14.4 Hz, J = 15.2 Hz, 1 H), 2.68 (ddd, JHP = 21.7 Hz, JHP = 12.9 Hz, J = 15.2 Hz, 1 H), 3.61–3.78 (m, 5 H), 4.08–4.41 (m, 5 H). 31P NMR (CDCl3, 121.47 MHz) δ 22.1 & 40.09 (2×d, JPP = 20.0 Hz).
Representative procedure for hindered iodides (Table 2, entry 8 ). Isopropyl-phenyl-phosphinic acid ethyl ester.20
Neat ethyl phenyl-H-phosphinate (510 mg, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. After 10 min, isopropyl iodide (300 μL, 3 mmol) was added under N2. After addition, the temperature of the solution was slowly allowed to warm to rt. The solution was then refluxed for 6 hours. After cooling, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc:Hexanes 80:20) to afford the desired product (45%). RN : [53716-14-6]. 1H NMR (CDCl3, 300 MHz) δ 1.04 (d, J = 7.0 Hz, 1.5 H), 1.10 (d, J = 7.0 Hz, 1.5 H), 1.16 (d, J = 7.0 Hz, 1.5 H), 1.22 (d, J = 7.0 Hz, 1.5 H), 1.32 (t, J = 7 Hz, 3 H), 2.0–2.15 (m, 1 H), 3.80–3.95 (m, 1 H), 4.05–4.20 (m, 1 H), 7.45–7.60 (m, 3 H), 7.70–7.80 (m, 2H). 31P NMR (CDCl3, 121.47 MHz) δ 54.60 (s).
Representative procedure with pyridinium salts (Table 3, entry 6b). (1,1-Diethoxy-ethyl)-pyridin-3-ylmethyl-phosphinic acid ethyl ester
Neat ethyl (l,l-diethoxyethyl)phosphinate (0.630 g, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. In a second dry two-neck flask, LiHMDS (1.0 M in THF, 3 mL, 3mmol) was added to a solution of 2-(bromomethyl)pyridine hydrobromide (0.759 g, 3 mmol) in anhydrous THF (5 mL), at −78°C under N2. After 10 min, the first solution was added to second one. After 10 min at −78°C, the temperature of the solution was slowly allowed to warm to rt. After 3 hours at rt, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc 100%) to afford the desired product (60%). 1H NMR (CDCl3, 300 MHz) δ 1.12–1.29 (m, 9 H), 1.50 (d, JHP = 11.4 Hz, 3 H), 3.11 & 3.23 (ABXsyst, JAB = 14.6 Hz, JBX = 8.2 Hz, JAX = 8.6 Hz, 2 H), 3.58–3.88 (m, 4 H), 4.08 (qt, J = 7.3 Hz, 2 H), 7.25 (dd, J = 7.9 Hz, J = 3.5 Hz, 1 H), 7.67–7.74 (m, 1 H), 8.48–8.51 (m, 2 H). 13C {1H} NMR (CDCl3, 75.45 MHz) δ 15.6 (d, JPOCC = 20.7 Hz), 16.7 (d, JPCC = 5.2 Hz), 20.6 (d, JPOCC = 12.4 Hz), 30.0 (d, JPC = 78 Hz), 57.9 (d, JPOC = 7.8 Hz), 58.7 (d, JPOC = 4.6 Hz), 62.3 (d, JPOC = 6.9 Hz), 101.5 (d, JPC = 142 Hz), 123.4, 127.3 (d, JPCC = 8.3 Hz), 137.8 (d, JPCCCC = 4.6 Hz), 148.2 (d, JPCCC = 3.2 Hz), 151.1 (d, JPCCCC = 6.0 Hz). 31P NMR (CDCl3, 121.47 MHz) δ 44.21 (s). HRMS (M+H ion by direct probe) : calc. for C14H25O4P 302.1521, obs. 302.1526.
Representative procedure with epoxides (Scheme 1). (2 - Hydroxy -hex-5-enyl)-phenyl-phosphinic acid ethyl ester 2
Neat ethyl phenyl-H-phosphinate (0.510 mg, 3 mmol) was placed under vacuum in a dry two-neck flask, during 10 min before use. Anhydrous THF (10 mL) was then added under nitrogen. The flask was placed at −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen and LiHMDS (1.0 M in THF, 3 mL, 3 mmol) was added at −78°C. After 10 min, 1,2-epoxy-5-hexene (340 μL, 3 mmol) was added followed by the addition of boron trifluoride etherate (380 μL, 3 mmol), under N2. After addition, the temperature of the solution was slowly allowed to warm to rt. After 2 hours at rt, the reaction mixture was quenched with NH4Cl/brine, and extracted with ethyl acetate (3 X) then dried over anhydrous MgSO4 and concentrated in vacuo. The resulting oil was purified by column chromatography (silica, EtOAc 100%) to afford the desired product (85%). 1H NMR (CDCl3, 300 MHz) δ 1.31 (t, J = 7.0 Hz, 3 H), 1.53–1.72 (m, 2 H), 1.91–2.19 (m, 5 H), 3.87 (m, 1 H), 4.07–4.23 (m, 2 H), 4.88–5.06 (m, 2 H), 5.78 (tqd, J = 6.4 Hz, J = 10.5 Hz, 1 H), 7.46–7.83 (m, 5 H). 13C {1H} NMR (CDCl3, 75.45 MHz) δ 16.7 (d, JPOCC = 6.6 Hz), 29.82, 36.2, 37.2 (d, JPC = 84.9 Hz), 37.5 (d, JPCCCC = 3.5 Hz), 37.6 (d, JPCCCC = 3.2 Hz), 61.1 (d, JPOCC = 6.9 Hz), 61.2 (d, JPOCC = 6.6 Hz), 65.6, 66.4 (d, JPOCC = 6.0 Hz), 128.9 (d, JPCC = 2.3 Hz), 129.1 (d, JPCC = 2.0 Hz), 130.4 (d, JPC = 128 Hz), 131.5 (d, JPCCC = 10.4 Hz), 131.8 (d, JPCCC = 10.1 Hz), 132.1, 132.8, 132.9, 138.2 (d, JPCCCC = 5.5 Hz). 31P NMR (CDCl3, 121.47 MHz) δ 44.26 (s), 55.74 (s). HRMS (EI+) : calc. for C14H21O3P 268.1228, obs. 268.1228.
Representative procedure with freon (Table 3, entry 9 ). Difluoromethyl-(1,1-diethoxy-ethyl)-phosphinic acid ethyl ester.1d
Neat ethyl (l,l-diethoxyethyl)phosphinate (12.0 g, 57.1 mmol) was placed under vacuum in a dry two-neck flask equipped with a cold finger, during 10 min before use. Anhydrous THF (80 mL) was then added under N2. The flask was cooled to −78°C and deoxygenated under vacuum for 5 min. The reaction flask was back-filled with nitrogen, then LiHMDS (1.0 M in THF, 57.1 mL, 57.1 mmol) was added at −78°C. After 15 min, condensed chlorodifluoromethane (around 5.0 g, 58.0 mmol) was added under N2. After addition, the temperature of the solution was kept at −78°C during 10 min, then slowly allowed to warm to 0°C. After 10 min at 0°C, the reaction mixture was quenched with a saturated solution of NH4Cl/brine, and extracted with EtOAc (3 X) then dried over anhydrous MgSO4. Concentration in vacuo gave an oil which was purified by column chromatography (silica, EtOAc:Hexanes 30:70) to afford the desired product (71%). RN : [139474-89-8]. 1H NMR (CDCl3, 300 MHz) δ 1.22 (t, J = 7.0 Hz, 6 H), 1.39 (t, J = 7.0 Hz, 3 H), 1.58 (d, JHP = 12.0 Hz, 3 H), 3.63–3.87 (m, 4 H), 4.29–4.39 (m, 2 H), 6.08 (dt, JHF = 27.5 Hz, JHF = 48.9 Hz, 1 H). 31P NMR (CDCl3, 121.47 MHz) δ 27.5 (t, JFP = 71.4 Hz). 19F NMR (CDCl3, 282.30 MHz) δ −135.31 (ddt, JFH = 21.8 Hz, JFP = 71.4 Hz, JFH = 49.5 Hz ).
Supplementary Material
Representative NMR spectra (98 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 2.

Synthesis of the GABA-B antagonist CGP 36742
Acknowledgments
The National Institute of General Medical Sciences/NIH (R01 GM067610) is gratefully acknowledged for financial support. JLM thanks Gerry Katchinska for a generous gift of CF2HCl, and Dr. Laëtitia Coudray for helpful discussions.
Footnotes
Alkylation of H-Phosphinate Esters
Fax: (+1) 817 257 5851, Phone: (+1) 817 257 6201, E-mail: j.montchamp@tcu
BRIEFS. The synthesis of differentially disubstituted phosphinic esters is conducted via alkylation of H-phosphinate esters under basic conditions.
In the case of the bromomethyl- or chloromethylpyridine hydrobromide or chloride, the pyridine was first deprotonated at −78C in dry THF with LiHMDS (1 eq.) under N2 for 15 min, then added to the solution of the lithium phosphinate.
The temperature and reaction time after the solution reaches rt depend on the reactivity of the electrophile.
References
- 1.Base-promoted alkylations of H-phosphinate esters: Baillie AC, Cornell CL, Wright BJ, Wright K. Tetrahedron Lett. 1992;33:5133. NaH.Baylis EK. Tetrahedron Lett. 1995;36:9385. (Na)Froestl W, Mickel SJ, von Sprecher G, Diel PJ, Hall RG, Maier L, Strub D, Melillo V, Baumann PA, Bernasconi R, Gentsch C, Hauser K, Jaekel J, Karlsson G, Klebs K, Maitre L, Marescaux C, Pozza MF, Schmutz M, Steinmann MW, van Riezen H, Vassout A, Mondadori C, Olpe HR, Waldmeier PC, Bittiger H. J Med Chem. 1995;38:3313. doi: 10.1021/jm00017a016. (NaH, BuLi)Froestl W, Mickel SJ, Hall RG, von Sprecher G, Diel PJ, Strub D, Baumann PA, Brugger F, Gentsch C, Jaekel J, Olpe HR, Rihs G, Vassout A, Waldmeier PC, Bittiger H. J Med Chem. 1995;38:3297. doi: 10.1021/jm00017a015. (NaH)Magnin DR, Biller SA, Dickson JK, Jr, Logan JV, Lawrence RM, Chen Y, Sulsky RB, Ciosek CP, Jr, Harrity TW, Jolibois KG, Kunselman LK, Rich LC, Slusarchyk DA. J Med Chem. 1995;38:2596. doi: 10.1021/jm00014a012. (NaHMDS)Gallagher MJ, Ranasinghe MG, Jenkins ID. Phosphorus, Sulfur, Silicon. 1996;115:255. (i-PrONa)Fairhurst RA, Collingwood SP, Lambert D, Taylor RJ. Synlett. 2001:467. (KHMDS)Larenco C, Villien L, Kaufmann G. Tetrahedron. 1984;40:2731. (NaH)McKittrick BA, Stamford AW, Weng X, Ma K, Chackalamannil S, Czarniecki M, Cleven RM, Fawzi AB. Bioorg Med Chem Lett. 1996;6:1629. (NaH, LDA)Froestl W, Bettler B, Bittiger H, Heid J, Kaupmann K, Mickel SJ, Strub D. Il Farmaco. 2003;58:173. doi: 10.1016/s0014-827x(03)00018-1. (NaH)Kehler J, Ebert B, Dahl O, Krogsgaard-Larsen P. Tetrahedron. 1999;55:771. (LDA, t-BuOK)Hemmi K, Takeno H, Hashimoto M, Kamiya T. Chem Pharm Bull. 1982;30:111. doi: 10.1248/cpb.30.111. (BuLi)Lindell SD, Turner RM. Tetrahedron Lett. 1990;31:5381. (NaH)Gallagher MJ, Honegger H. Tetrahedron Lett. 1977:2987. (MeONa)Hall RG, Kane PD, Bittiger H, Froestl W. J Label Compounds Radiopharm. 1995:129.
- 2.For some representative examples of the silicon method, see: Boyd EA, Regan AC, James K. Synthesis of Alkyl Phosphinic Acids from Silyl Phosphonites and Alkyl Halides. Tetrahedron Lett. 1994;35:4223.Boyd EA, Corless M, James K, Regan AC. A Versatile Route to Substituted Phosphinic Acids. Tetrahedron Lett. 1990;31:2933.Malachowski WP, Coward JK. J Org Chem. 1994;59:7625.Reck F, Marmor S, Fisher S, Wuonola MA. Bioorg Med Chem Lett. 2001;11:1451. doi: 10.1016/s0960-894x(01)00251-7.Ribière P, Altamirano-Bravo K, Antczak MI, Hawkins JD, Montchamp JL. J Org Chem. 2005;70:4064. doi: 10.1021/jo050096l. See also references 1c, 1d, 1h
- 3.Representative examples: Grembecka J, Mucha A, Cierpicki T, Kafarski P. J Med Chem. 2003;46:2641. doi: 10.1021/jm030795v.Lloyd J, Schmidt JB, Hunt JT, Barrish JC, Little DK, Tymiak AA. Bioorg Med Chem Lett. 1996;6:1323.Karanewsky DS, Badia MC, Cushman DW, DeForrest JM, Dejneka T, Loots MJ, Perri MG, Petrillo EW, Jr, Powell JR. J Med Chem. 1988;31:204. doi: 10.1021/jm00396a033.Qiao L, Nan F, Kunkel M, Gallegos A, Powis G, Kozikowski AP. J Med Chem. 1998;41:3303. doi: 10.1021/jm980254j.Tokutake N, Hiratake J, Katoh M, Irie T, Kato H, Oda J. Bioorg Med Chem. 1998;6:1935. doi: 10.1016/s0968-0896(98)00142-4.Manthey MK, Huang DTC, Bubb WA, Christopherson RI. J Med Chem. 1998;41:4550. doi: 10.1021/jm970814z.Ebetino FH, Berk JD. J Organomet Chem. 1997;529:135.Vayron P, Renard PY, Valleix A, Mioskowski C. Chem Eur J. 2000;6:1050. doi: 10.1002/(sici)1521-3765(20000317)6:6<1050::aid-chem1050>3.0.co;2-5.Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzati BA, Trainor DA. J Med Chem. 1996;39:619. doi: 10.1021/jm950801q.Hiratake J. Chemical Record. 2005;5:209. doi: 10.1002/tcr.20045.Bartley DM, Coward JK. J Org Chem. 2005;70:6757. doi: 10.1021/jo0507439.Valiaeva N, Bartley D, Konno T, Coward JK. J Org Chem. 2001;66:5146. doi: 10.1021/jo010283t.Jackson PF, Tays KL, Maclin KM, Ko YS, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu XCM, Wozniak K, Slusher BS. J Med Chem. 2001;44:4170. doi: 10.1021/jm0001774.Collinsova M, Jiracek J. Curr Med Chem. 2000;7:629. doi: 10.2174/0929867003374831.Flohr A, Aemissegger A, Hilvert D. J Med Chem. 1999;42:2633. doi: 10.1021/jm991008q.Chen S, Coward JK. J Org Chem. 1998;63:502. doi: 10.1021/jo971318l.Hiratake J, Kato H, Oda J-i. J Am Chem Soc. 1994;116:12059.
- 4.Reviews: Montchamp JL. J Organomet Chem. 2005;690:2388.Montchamp JL. Specialty Chemicals Magazine. 2006;26:44.
- 5.Other workers have used deoxygenation previously, see ref. 1k.
- 6.For applications of the “Ciba-Geigy reagents”, see: Dingwall JG, Ehrenfreund J, Hall RG, Jack J. Phosphorus Sulfur. 1987;30:571.McCleery PP, Tuck B. J Chem Soc Perkin Trans I. 1989:1319.Dingwall JG, Ehrenfreund J, Hall RG. Tetrahedron. 1989;45:3787.Baylis EK. Tetrahedron Lett. 1995;36:9385.Baylis EK. Tetrahedron Lett. 1995;36:9389.Bennett SNL, Hall RG. J Chem Soc Trans 1. 1995:1145. See also refs 1c and 1d
- 7.Cleavage of ketal protecting group to H-phosphinate: see ref. 6.
- 8.Abrunhosa-Thomas I, Ribière P, Adcock AC, Montchamp JL. Synthesis. 2006;2:325. [Google Scholar]
- 9.Gallagher MJ, Ranasinghe MG, Jenkins ID. Phosphorus, Sulfur, Silicon. 1996;115:255. [Google Scholar]
- 10.The compounds were inactive.
- 11.CH3C(OEt)2P(O)(OEt)CF2H has been prepared in 95% yield using NaH, and was used in situ: see ref 1c. The synthetic use of this and other difluorophosphinates described herein will be reported separately.
- 12.For examples of epoxide-opening using the silicon method, see: refs 1c, 1k.
- 13.CGP36742/SGS742: Chebib M, Vandenberg RJ, Froestl W, Johnston GAR. Eur J Pharmacol. 1997;329:223.Pittaluga A, Vaccari D, Raiteri M. J Pharmacol Exp Ther. 1997;283:82.Steulet AF, Moebius HJ, Mickel SJ, Stoecklin K, Waldmeier PC. Biochem Pharmacol. 1996;51:613. doi: 10.1016/s0006-2952(95)02183-3.
- 14.Deprèle S, Montchamp JL. J Org Chem. 2001;66:6745. doi: 10.1021/jo015876i. [DOI] [PubMed] [Google Scholar]
- 15.Dumond YR, Baker RL, Montchamp JL. Org Lett. 2000;2:3341. doi: 10.1021/ol006434g. [DOI] [PubMed] [Google Scholar]
- 16.Ross FC, Botting NP, Leeson PD. Bioorg Med Chem Lett. 1996;6:2643. [Google Scholar]
- 17.(a) Zeiss HG. J Org Chem. 1991;56:1783. [Google Scholar]; (b) Maier L, Rist G. Phosphorus, Sulfur. 1983;17:21. [Google Scholar]; (c) Tan S, Evans R, Singh B. Amino Acids. 2006;30:195. doi: 10.1007/s00726-005-0254-1. [DOI] [PubMed] [Google Scholar]; (d) Evstigneeva ZG, Solov’eva NA, Sidel’nikova LI. Appl Biochem Microbiol. 2003;39:539. [Google Scholar]
- 18.Pudovik AN, Konovalova IV. J Gen Chem USSR. 1960;30:2328. [Google Scholar]
- 19.Luke GP, Shakespeare WC. Synth Commun. 2002;32:2951. [Google Scholar]
- 20.(a) Zymanczyk-Duda E, Lejczak B, Kafarski P. Phosphorus, Sulfur Silicon Relat Elem. 1996;112:47. [Google Scholar]; (b) Siddall TH, III, Prohaska CA. J Am Chem Soc. 1962;84:2502. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative NMR spectra (98 pages). This material is available free of charge via the Internet at http://pubs.acs.org.