

Abstract

A de novo approach to the formal total synthesis of the macrolide natural product (—)-virginiamycin M2 has been achieved via a convergent approach. The absolute and relative stereochemistry of the non-peptide portion of (—)-virginiamycin M2 was introduced by two Sharpless asymmetric dihydroxylation reactions.
The problem associated with new microbes that develop resistance mechanisms to antibiotics has fueled the never-ending search for new antibacterial agents.1 In fact, infections from methicillin-resistant S. aureus (MRSA) organism have become increasingly common.2 More alarming is the discovery of vancomycin resistant S. aureas organisms.3 Currently three antibiotics (linezolid, daptomycin, and quinupristin-dalfopristin) have been approved to combat these infections. The oldest of the three quinupristin-dalfopristin is the admixture of two types of streptogramin antibiotics, dalfopristin (type A) and quinupristin (type B) (Figure 1), which derives its potency by the synergistic binding of two weak ribosome binders.1
Figure 1.
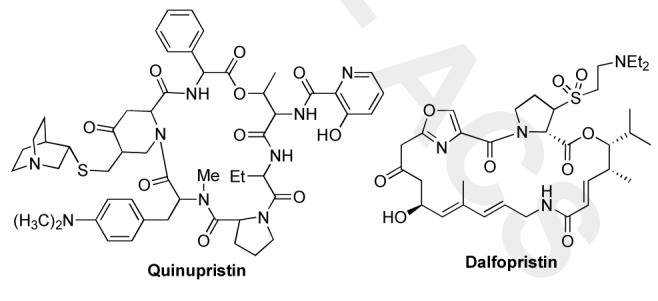
Quinupristin-dalfopristin
The type A component dalfopristin is a semi-synthetic compound prepared by a sulfinic acid addition across the dehydro-proline portion of virginiamycin M1 (Figure 2), which enhances activity by providing improved solubility.1c As part of a program aimed at developing new antibiotic structures we became interested in the synthesis of both virginiamycin M2 as well as the synthesis of a library of novel virginiamycin analogues. We envisioned replacing the sulfone group on the proline with an aminoglycoside sugar, with the hope of discovering new synergistic binding.4 As a prelude to this medicinal chemistry studies/library synthesis, we decided to investigate the feasibility by conducting a formal total synthesis.
Figure 2.
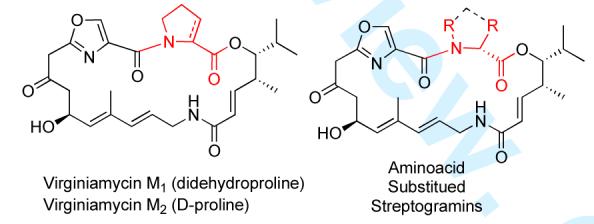
Type A Streptogramins
In addition to its potent antibiotic activity, the structural novelty of virginiamycin M2 has also attracted the attention of the synthetic community.5,6 To date several total syntheses of the type A streptogramin antibiotics have been completed,5 along with a formal total synthesis.6 While all of the previous syntheses of the streptogramin class of antibiotics derived their asymmetry from the chiral pool, enzymatic resolution and/or chiral auxilaries,5 we were interested in a de novo asymmetric approach that would use asymmetric catalysis to install the stereocenters of the nonaminoacid portion of type A streptogramins. Herein we describe our successful efforts to implement this strategy for the de novo formal total synthesis of virginiamycin M2.
Retrosynthetically, virginiamycin M2 (1) has been derived from the seco-macrolide 2,5b which in turn could be prepared from the known oxazole 3, and triene 4. Previously, Schlessinger had demonstrated the conversion of 4 and 3 to Virginiamycin M2 (1).5a In our strategy (Scheme 1), we envisioned the triene 4 as being assembled from D-proline, allylic amine 5 and δ-hydroxy ester 6. Finally we planed to install the asymmetry of these two fragments by the application of a regioselective Sharpless asymmetric dihydroxylation of diene fragments 7 and 8.7,8 In particular, we were interested in using our asymmetric hydration strategy for the preparation of the syn-γ-substituted δ-hydroxyenoate 6.9
Scheme 1.
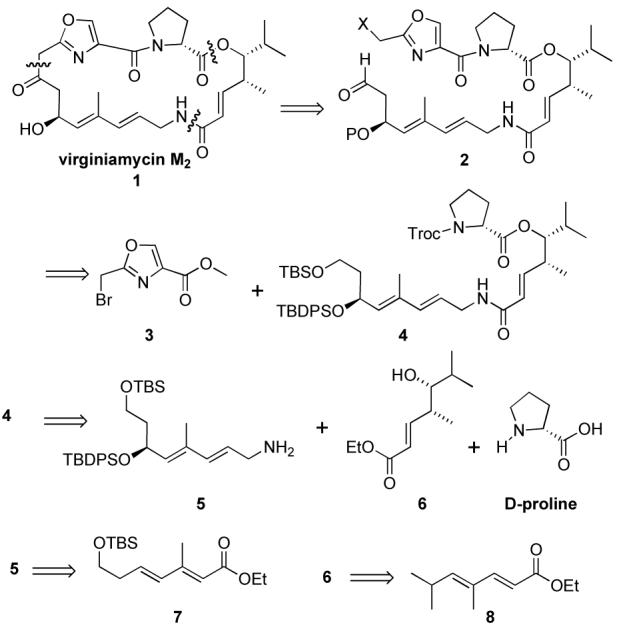
Retrosynthesis of (—)-Virginiamycin M2 (1)
To access useful quantities of dienoate 8, an efficient 5-step approach was developed (Scheme 2). While the route featured standard Wittig/Horner Emmons olefination chemistry, key to practical nature of this approach was the recognition that TFA can catalyze the stereoselective isomerization of enal 11 to the more stable E-isomer (98:2), which when treated with the stablized Wittig reagent provided good yields of 8 (72%).10 While we previously had demonstrated the asymmetric hydration of 8 to the enantiomer of 6, because of the pseudo-enantiomeric nature of the Sharpless reagents (DHQ/DHQD) the regiochemistry of this reaction was an open question. In practice, we found that the monomeric 4-methyl-2-quinolylether linked DHQD ligand provided the best balance in terms of regio- and stereochemistry.11 Thus when 8 was dihydroxylated with the OsO4/DHQD-4-MEQ reagent good yields of diol 12 as a single regioisomer was isolated (75%, 90% ee), which was cyclized into carbonate13 in good overall yield (90%). Exposure of carbonate 13 to the palladium(0) catalyzed reduction conditions (HCO2H/Et3N) provided δ-hydroxy enoate 6 in good yield (98%).
Scheme 2.
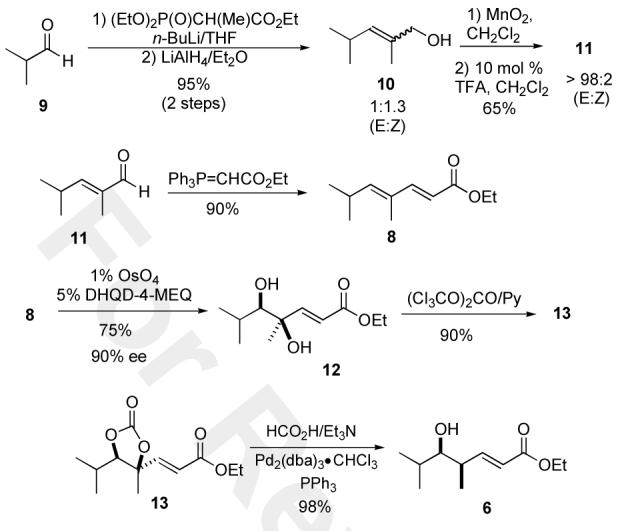
Asymmetric hydration of dienoate8
With δ-hydroxyenoate 6 in hand, we next investigated the synthesis of amine 5 (Scheme 3). Unfortunately, our initial plan to use a regioselective asymmetric dihydroxylation of dienoate 7, was thwarted by our inability to prepare 7 by Horner-Emmons olefination of 15. Instead of 7 only Michael addition products were observed. Thus, we decided to preform the dihydroxylation a step earlier on enone 15, which could be easily prepared in three steps (63%) from 1,3-propane diol (14). Exposure of enone 15 to the typical Sharpless asymmetric dihydroxylation procedures gave a good yield (71%) of diol 16 and in high enantiopurity (90% ee). The diol product was protected as an acetonide (2,2-DMP/5% CSA, 72%) and the ketone product 17 underwent Horner-Emmons olefination to form enoate 18 (84%; 10:1, E/Z). The enoate 18 was reduced with DibalH to give allylic alcohol 19, which in turn was converted into allylic nitrile 20 by a 2-step protocol (PPh3/I2 then NaCN in AN; 50%). Base promoted elimination (K2CO3/MeOH, 81%) of 20 stereoselectively gave the E,E-diene 21 (98:2, EE:EZ). The secondary allylic alcohol in 21 was protected as a TBDPS-ether (TBDPSCl/imid, 89%) and the nitrile was cleanly reduced with AlH3 to give amine 5 in good yield (70%).12
Scheme 3.
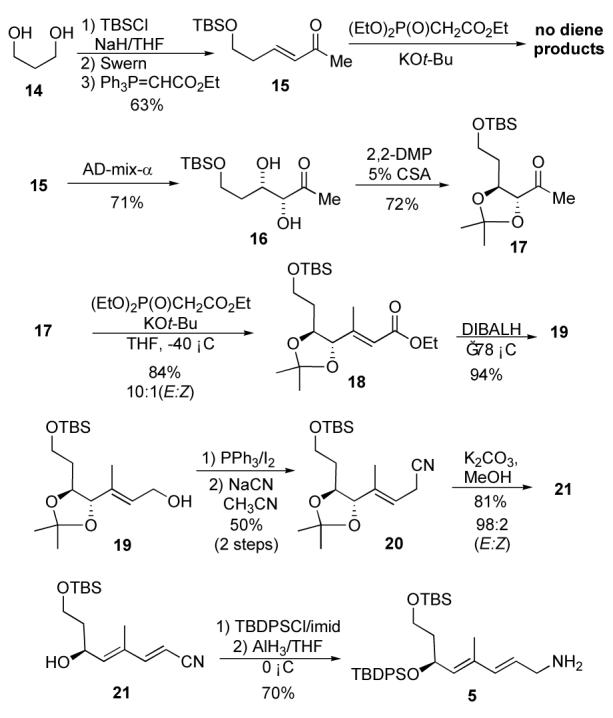
De novo synthesis of allylic amine 5
We next investigated the synthesis of the first fragment coupling product ester 25 (Scheme 4). While 24 could be prepared in one step from a Troc-protected D-proline, for this model system, we decided to use the already available Boc-protected D-proline 22. Coupling proline 22 and alcohol 6 with DCC/DMAP followed by deprotection of the Boc-group (TFA) gave good yields of amine 23 (88%, 2 steps). The secondary amine 23 was protected with TrocCl/Pyr (71%) to give ester 24, which was selectively hydrolyzed (NaOH, 56%) to give the desired carboxylic acid 25.13
Scheme 4.
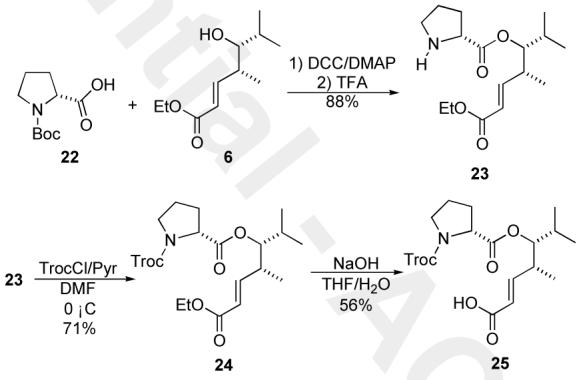
Synthesis of proline ester 25
With the final two fragments 5 and 25 in hand, we investigated their coupling to form our desired target molecule 4 (Scheme 5). After screening several coupling procedures we found the DCC/DMAP to give the best yields and to be operationally the simplest. Thus, exposing a 1:1 mixture of 5 and 25 to a CH2Cl2 solution of DCC/DMAP gave 82% yield of amide 4, which was physically (optical rotation) and spectroscopically (1H NMR, 13C NMR, IR and MS) identical to the material previously reported by Schlessinger.14
Scheme 5.
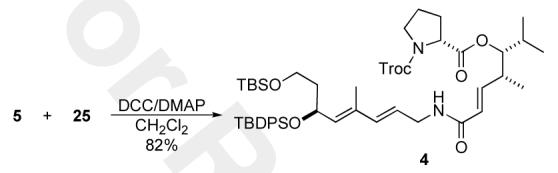
Completion of the formal synthesis of (—)-virginiamycin M2
In conclusion, a short formal de novo asymmetric synthesis of virginiamycin M2 has been developed. This highly enantio- and diastereocontrolled route illustrates the utility of our dienoate asymmetric hydration strategy for natural product synthesis. Further application of this approach to the synthesis of mixed aminoglycoside/virginiamycin M2 analogues is ongoing and these results will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM63150) and NSF (CHE-0415469) for the support of our research program and NSF-EPSCoR (0314742) for a 600 MHz NMR and an LTQ-FT Mass Spectrometer at WVU.
Footnotes
Supporting Information Available: Complete experimental procedures and spectral data for all new compounds can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walsh CT. Nature. 2000;406:775–781. doi: 10.1038/35021219. Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ, Cardo D, Fridkin SK. N. Engl. J. Med. 2003;348:1342–1349. doi: 10.1056/NEJMoa025025. and refs therein.Manthous CA, Amaoteng-Adjepong Y. Chest. 2000;118:9–11. doi: 10.1378/chest.118.1.9.For clinical and pharmacological background on streptomycin antibiotics see:Barriere JC, Berthaud N, Beyer D, Dutka Malen, S., Paris JM, Desnottes JF. Curr. Pharm. Des. 1998;4:155–180.Manzella JP. Am. Fam. Physician. 2001;64:1863–1866.Mukhtar TA, Wright GD. Chem. Rev. 2005;105:529–542. doi: 10.1021/cr030110z.Porse BT, Garrett RA. J. Mol. Biol. 1999;286:375. doi: 10.1006/jmbi.1998.2509.For synthesis of new quinolone-class antibiotic active against VRSA see:de Souza NJ, Gupte SV, Deshpande PK, Desai VN, Bhawsar SB, Yeole RD, Shukla MC, Strahilevitz J, Hooper DC, Bozdogan B, Applebaum PC, Jacobs MR, Shetty N, Patel MV, Jha R, Khorakiwala HF. J. Med. Chem. 2005;48:5232–5242. doi: 10.1021/jm050035f.
- 2.McCaughey B. N. Y. Times. 2006;(November 14):27. [Google Scholar]
- 3.Schito GC. Clin Microbiol Infect. 2006;12:3–8. doi: 10.1111/j.1469-0691.2006.01343.x. [DOI] [PubMed] [Google Scholar]
- 4 (a).For conformational studies of the virginiamycin type antibiotics see: Dang J, Metzger RP, Brownlee RTC, Chai AN, Separovic F. The Conformation Flexibility of the Antibiotic Virginiamycin M1. Eur. J. Biophys. 2005;34:383–388. doi: 10.1007/s00249-005-0464-1.Dang J, Separovic F, Brownlee RTC, Metzger RP. J. Org. Biomol. Chem. 2004;2:2919–2924. doi: 10.1039/B407724E.Dang J, Separovic F, Brownlee RTC, Metzger RP. Aust. J. Chem. 2004;57:415–418.
- 5 (a).For the first total synthesis of virginiamycin M2, see: Schlessinger RH, Li Y-J. J. Am. Chem. Soc. 1996;118:3301–3302. For the second see:Breuilles P, Uguen D. Tetrahedron Lett. 1998;39:3149–3152.For the first total synthesis of madumycin II, see:Tavares F, Lawson JP, Meyers AI. J. Am. Chem. Soc. 1996;118:3303–3304. For the second see:Ghosh AK, Liu W. J. Org. Chem. 1997;62:7908–7909. doi: 10.1021/jo971616i.
- 6.Brennan CJ, Campagne J-M. Tetrahedron Lett. 2001;42:5195–5197. [Google Scholar]
- 7 (a).The regioselectivity of the asymmetric dihydroxylation of di- and trienoates has been studied by Sharpless, see: Berker H, Soler MA, Sharpless KB. Tetrahedron. 1995;51:1345–1376.and our group, see:Zhang Y, O’Doherty GA. Tetrahedron. 2005;61:6337–6351.
- 8 (a).For the use of this approach in synthesis, see: Smith AB, Walsh SP, Frohn M, Duffey MO. Org. Lett. 2005;7:139–142. doi: 10.1021/ol047792c.Ahmed Md. M., Akhmedov N, Cui H, Friedrich D, O’Doherty GA. Heterocycles. 2006;70:223–233.Ahmed Md. M., Cui H, O’Doherty GA. J. Org. Chem. 2006;71:6686–6689. doi: 10.1021/jo061057s.Gao D, O'’Doherty GA. J. Org. Chem. 2005;70:9932–9939. doi: 10.1021/jo051681p.Ahmed Md. M., O’Doherty GA. Tetrahedron Lett. 2005;46:4151–4155.
- 9 (a).Ahmed Md. M., Mortensen MS, O’Doherty GA. J. Org. Chem. 2006;71:7741–7746. doi: 10.1021/jo061200h.Hunter TJ, O’Doherty GA. Org. Lett. 2001;3:1049–1052. doi: 10.1021/ol0156188.For its use in the synthesis of natural products see:Li M, O’Doherty GA. Org. Lett. 2006;8:3987–3990. doi: 10.1021/ol061439k. andLi M, O’Doherty GA. Org. Lett. 2006;8:6087–6090. doi: 10.1021/ol062595u.
- 10.We have found this acid catalyzed isomerization of α,β-unsaturated aldehydes to be quite general and very useful in our diene syntheses, see: Varelis P, Johnson B. Aust. J. Chem. 1997;50:43–52.
- 11.In addition to the electronic effect, we have found subtle steric effects can lead to the formation of regioisomers in the Sharpless asymmetric dihydroxylation reaction of dienoates, see refs 7 and 9a.
- 12.The chemoselective AlH3 reduction of dienylnitriles like 21 is precedented, see ref. 5a.
- 13.We surmised that this lower than expected yield was due to the water solubility of 25, no elimination to dienoate 8 was observed.
- 14.Unfortunately a typographical error occurs in the data reported by Schlessinger such that the 11 signals below 24 ppm are missing from their reported 13C NMR spectral data. The remaining 25 signals above 24 ppm did match the reported data, see ref. 5a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.