

Abstract
 This manuscript describes a highly site, diastereo-, and enantioselective intermolecular radical addition/hydrogen atom transfer to hydroxypyrones pyromeconic and kojic acids. The methodology can be extended to the formation of chiral quaternary centers. The products obtained are densely functionalized pyran moieties. The products contain structural features amenable for the introduction of additional substituents.
This manuscript describes a highly site, diastereo-, and enantioselective intermolecular radical addition/hydrogen atom transfer to hydroxypyrones pyromeconic and kojic acids. The methodology can be extended to the formation of chiral quaternary centers. The products obtained are densely functionalized pyran moieties. The products contain structural features amenable for the introduction of additional substituents.
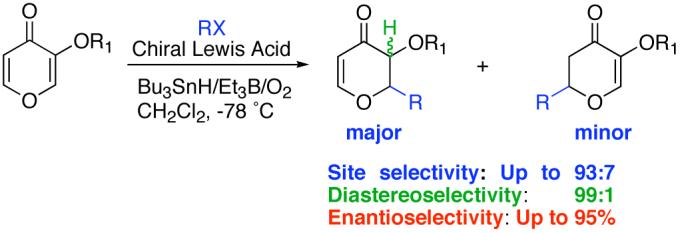
Enantioselective Lewis-acid mediated free radical reactions continue to attract interest.1 Detachable achiral auxiliaries play a pivotal role in most of the enantioselective radical chemistry reported in the literature to date.2 These auxiliaries provide an extra donor atom that enables Lewis acid chelation. Another significant structural feature is that, in general, α,β-unsaturated carbonyl compounds that have undergone enantioselective radical additions have reacted via s-cis conformers.
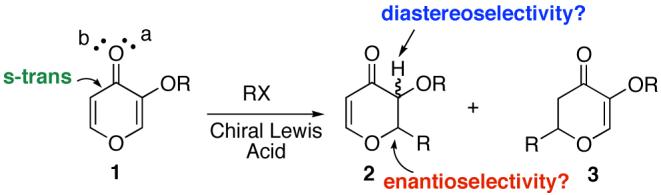
We have been interested in developing enantioselective radical additions onto substrates that will not require an achiral template3 and react via an s-trans geometry. Stereocontrolled functionalization of readily available hydroxypyrones4 is important since they provide access to pyrans, a structural unit present in compounds with significant biological activity. For example, marine natural products apicularen, phorboxazole and spongistatin all contain pyran rings with various substitution patterns.5 Recently, Hoveyda has demonstrated enantioselective conjugate additions to chromones.4e This communication addresses several challenging issues for Lewis acid catalyzed conjugate radical additions to hydroxypyrones (Scheme 1): (1) how reactive are they towards conjugate radical additions? (2) what is the C-2 vs. C-6 site selectivity (2 vs 3) and will electronic or steric factors act as control elements? (3) what will be the optimal chiral Lewis acid system for stereoselectivity?
Scheme 1.
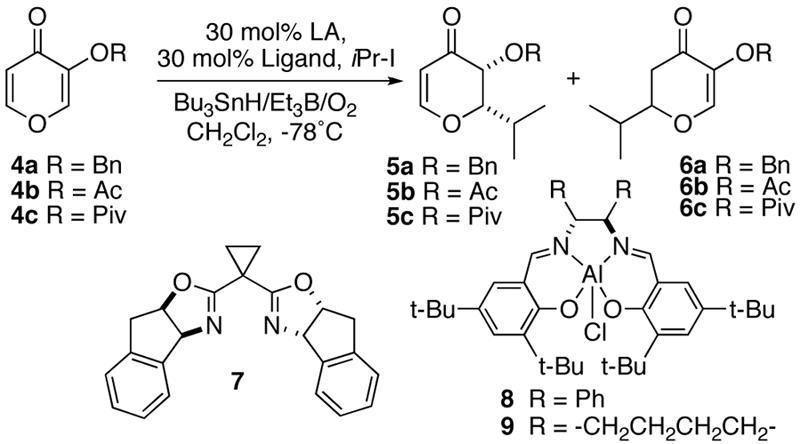
Our initial studies began using pyromeconic acid derivatives 4 (Table 1). It was found early on that the enol hydroxyl group needed to be functionalized with an electron-withdrawing group in order to achieve good reactivity (entry 1). No reaction took place (<5%) for substrates 4a-c without a Lewis acid. More interesting was the effect of the Lewis acid in these reactions. Initial racemic reactions showed that magnesium and scandium salts yielded a mixture of isomers (entries 2-5).6 Traditional bisoxazoline ligands were screened with magnesium and scandium Lewis acids but only low to moderate enantioselectivities were achieved (entries 6-8). Promising results were obtained using chiral aluminum salen derived catalysts (entries 9-11).7 The commercially available 9 gave 5c in excellent yield and diastereoselectivity with the major isomer possessing syn stereochemistry.8 The C-2 isomer was preferentially formed (≥10:1) in the Al-salen catalyzed reactions with moderate enantioselectivities (entry 11).9
Table 1.
Lewis Acid Screening with Pyromeconic Acid Derivatives
 | |||||||
|---|---|---|---|---|---|---|---|
| Ent | R | Lewis Acid |
Lig. | Yield %b |
dr (5)c syn/anti |
5:6 | ee (5) (%) |
| 1 | Bn | Sc(OTf)3 | - | trace | - | - | - |
| 2 | Ac | Mg(NTf2)2 | - | 22 | 95:5 | 3.3:1 | - |
| 3 | Ac | Sc(OTf)3 | - | 78 | 86:14 | 1.9:1 | - |
| 4 | Piv | Mg(NTf2)2 | - | 60 | 99:1 | 5.3:1 | - |
| 5 | Piv | Sc(OTf)3 | - | 76 | 91:9 | 2.6:1 | - |
| 6 | Piv | Mg(NTf2)2 | 7 | 40 | 91:9 | 1.1:1 | 62 |
| 7 | Piv | Mg(ClO4)2 | 7 | 33 | 99:1 | 4.0:1 | 16 |
| 8 | Piv | Sc(OTf)3 | 7 | 80 | 88:12 | 4.5:1 | 3 |
| 9 | Piv | 8 | 8 | 71 | 99:1 | 13:1 | 30 |
| 10 | Ac | 9 | 9 | 60 | 99:1 | 4.0:1 | 49 |
| 11 | Piv | 9 | 9 | 80 | 99:1 | 10:1 | 70 |
For reaction conditions see supporting information.
Isolated yield.
Diastereomer ratio and site selectivity (5:6) were determined by 1H NMR (400 MHz).
Determined by chiral HPLC.
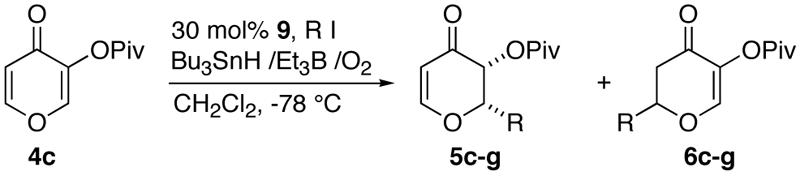
We next investigated the addition of various radicals to substrate 4c under the optimized conditions (Table 2). Addition of secondary radicals gave good yields, excellent diastereoselectivity, and moderate enantioselectivity for the major C-2 product (5) (entries 1-3). The minor C-6 products (6) were analyzed in two cases and found to be much less selective with ee's less than 40% (entries 2 and 5). In contrast, reactions with bulky tertiary radicals yielded the major C-2 adducts with excellent diastereo- and enantioselectivities (>90%) (entries 4-6). Another important point of note is that lowering the catalytic loading to 30 mol% showed no erosion of enantioselectivity (compare entry 4 with 5).
Table 2.
Radical Additions to Pyromeconic Acid Pivalate
 | |||||
|---|---|---|---|---|---|
| Ent | R1 | 5:6 | Yld, %a | drb | ee, % 5 (6)c |
| 1 | i-Propyl 5c | 91:9 | 80 | 99:1 | 70 |
| 2 | c-Pentyl 5d | 93:7 | 74 | 99:1 | 76 (36) |
| 3 | c-Hexyl 5e | 92:8 | 55 | 99:1 | 70 |
| 4 | t-Butyl 5f | 90:10 | 76 | 99:1 | 92 |
| 5d | t-Butyl 5f | 93:7 | 60 | 99:1 | 92 |
| 6 | (CH3)2C(CH2)3Cl 5g | 92:8 | 45 | 99:1 | 95 (39) |
Isolated yield.
Diastereomer ratio and site selectivity (5:6) were determined by 1H NMR (400 MHz).
Determined by chiral HPLC.
100 mol% 9.
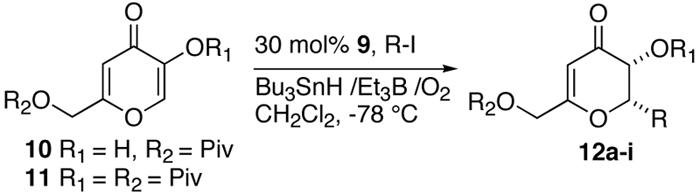
We next focused our attention on kojic acid, which is an inexpensive, commercially available starting pyrone. In this case the C-6 position is functionalized with a hydroxymethyl moiety, which could provide a handle for further synthetic manipulations but also deactivates the C-6 position toward conjugate radical addition. Kojic acid is relatively insoluble in nonpolar solvents and thus was converted to 10-11 to increase solubility. Again, no reaction took place without Lewis acid for substrates 10 and 11 (Table 3). As was expected, radical addition occurs exclusively at the less substituted C-2 position. Addition of secondary radicals to 10 proceeded smoothly to yield 12a-b in good yields, excellent diastereoselectivity and moderate ee's (entry 1 and 2). More bulky tertiary radicals again proved to give enantioselectivities >90% (entries 3 and 4). We also examined the bis-pivalyl substrate 11. Again excellent yields and diastereoselectivities were obtained when secondary nucleophilic radicals were added, while tertiary radicals gave excellent ee's (entries 5-9).
Table 3.
Radical Addition to Kojic Acid Derivatives
 | ||||||
|---|---|---|---|---|---|---|
| Ent | R | R1 | R2 | Yield (%)a |
drb | ee (%)c |
| 1 | i-Propyl 12a | H | Piv | 85 | 99:1 | 74 |
| 2 | c-Pentyl 12b | H | Piv | 67 | 99:1 | 76 |
| 3 | t-Butyl 12c | H | Piv | 91 | 99:1 | 92 |
| 4 | (CH3)2C(CH2)3Cl 12d | H | Piv | 35 | 99:1 | 92 |
| 5 | i-Propyl 12e | Piv | Piv | 92 | 99:1 | 72 |
| 6 | c-Pentyl 12f | Piv | Piv | 74 | 99:1 | 76 |
| 7 | t-Butyl 12g | Piv | Piv | 96 | 99:1 | 93 |
| 8d | t-Butyl 12g | Piv | Piv | 98 | 99:1 | 92 |
| 9 | (CH3)2C(CH2)3Cl 12h | Piv | Piv | 77 | 99:1 | 90 |
Isolated yield.
Diastereomer ratio determined by 1H NMR (400 MHz).
Determined by chiral HPLC.
10 mol% 9.
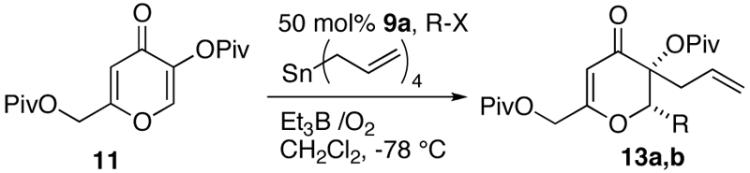
We were also interested in forming two carbon-carbon bonds via conjugate addition of a nucleophilic radical followed by trapping of the electrophilic α-radical with an allyltin reagent.10 This process establishes two new adjacent stereocenters with one being a quaternary carbon. Initial attempts using catalyst 9 were unsuccessful, but by utilizing a more reactive chiral Lewis acid (Cl exchanged to NTf2, 9a)11 we were able to obtain good to excellent yields of the addition/trapping products 13a,b (Table 4).12 The enantioselectivities are modest and similar to what was observed under reductive tin hydride conditions for isopropyl radical addition (Table 3).
Table 4.
Quaternary Center Formation
Isolated yield.
Diastereomer ratio determined by 1H NMR (400 MHz).
Determined by chiral HPLC.
The absolute stereochemistry for product 5e was determined by conversion to a known compound.13 Figure 1 shows a proposed chiral Lewis acid-substrate model which accounts for the observed stereochemistry in pyromeconic acid radical conjugate additions. We propose that the substrate binds through the ketone carbonyl to the Al-salen catalyst.14 The electron-withdrawing acyloxy substituent may facilitate addition at C-2, even though C-6 is more sterically accessible. Captodative effects may also impact regioselectivity. The bulky –OR group orients away from the axial hydrogen atoms on the cyclohexane ring leaving the si-face more open for nucleophilic radical addition. Subsequent hydrogen transfer to the αcarbon is apparently controlled not by the chiral ligand, but by the newly formed βstereocenter, with the radical R group shielding the top face.
Figure 1.
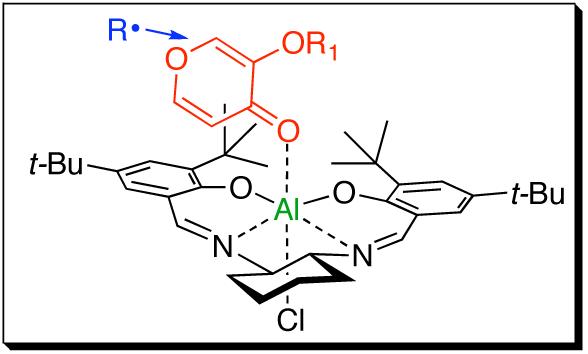
Stereochemical model
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (NIH-GM-54656).
Footnotes
Supporting Information Available: Characterization data for compounds 4-13 and experimental procedures. See any current masthead page for ordering information and Web access instructions.
REFERENCES
- 1.(a) Sibi MP, Manyem S, Zimmerman J. Chem. Rev. 2003;103:3263. doi: 10.1021/cr020044l. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP, Porter NA. Acc. Chem. Res. 1999;33:163. [Google Scholar]; Sibi MP, Manyem S. Tetrahedron. 2000;56:8033. [Google Scholar]
- 2.(a) Sibi MP, Sausker JB. J. Am. Chem. Soc. 2002;124:984. doi: 10.1021/ja016839b. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP, Prabagaran N. Synlett. 2004:2421. [Google Scholar]; (c) Sibi MP, Shay JJ, Ji J. Tetrahedron Lett. 1997;38:5955. [Google Scholar]
- 3.(a) Sibi MP, Patil K. Angew. Chem. Int. Ed. 2004;43:1235. doi: 10.1002/anie.200353000. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP, Patil K. Org. Lett. 2005;6:1543. doi: 10.1021/ol047347h. [DOI] [PubMed] [Google Scholar]; (c) Sibi MP, Asano Y, Sausker JB. Angew. Chem. Int. Ed. Engl. 2001;40:1293. doi: 10.1002/1521-3773(20010401)40:7<1293::aid-anie1293>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 4.Selected examples on the use of pyrones in synthesis see: Woodard BT, Posner GH. Adv. In Cycloaddition. 1999;5:47.Marko IE, Evans GR, Seres P, Chelle I, Janousek Z. Pure App. Chem. 1996;68:113.Wender PA, McDonald FE. J. Am. Chem. Soc. 1990;112:4956.Rodriguez JR, Rumbo A, Castedo L, Mascarenas JL. J. Org. Chem. 1999;64:4560. doi: 10.1021/jo982050g.Brown MK, Degrado SJ, Hoveyda AH. Angew. Chem. Int. Ed. 2005;44:5306. doi: 10.1002/anie.200501251.Okamura H, Iwagawa T, Nakatani M. Tetrahedron Lett. 1995;36:5939.
- 5.(a) Smith AB, III, Adams CM. Acc. Chem. Res. 2004;37:365. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]; (b) Su Q, Panek JS. J. Am. Chem. Soc. 2004;126:2425. doi: 10.1021/ja037957x. [DOI] [PubMed] [Google Scholar]; (c) Haustedt LO, Hartung IV, Hoffmann HMR. Angew. Chem. Int. Ed. 2003;42:2711. doi: 10.1002/anie.200301643. [DOI] [PubMed] [Google Scholar]
- 6.For the synthesis of starting materials, reaction conditions for radical reactions, ee determination, and product stereochemical analysis see supporting information.
- 7.Cozzi PG. Chem. Soc. Rev. 2004;33:410. doi: 10.1039/b307853c. For a review on salen catalysts in synthesis see:Selected examples on the use of salens in conjugate additions, see: Myers JK, Jacobsen EN. J. Am. Chem. Soc. 1999;121:8959.Taylor MS, Zalatan DB, Lerchner AM, Jacobsen EN. J. Am. Chem. Soc. 2005;127:1313. doi: 10.1021/ja044999s.
- 8.The relative stereochemistry was assigned by analogy of a very similar series of compounds, see: Yamashita Y, Saito S, Ishitani H, Kobayashi S. J. Am. Chem. Soc. 2003;125:3793. doi: 10.1021/ja028186k.
- 9.For site selectivity in radical addition to 2-methoxybenzoquinone see: Ling T, Poupon E, Rueden EJ, Kim SH, Theodorakis EA. J. Am. Chem. Soc. 2002;124:12261. doi: 10.1021/ja027517q.
- 10.Rosenstein IJ. In: Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 1. Wiley-VCH; New York: 2001. pp. 50–71. [Google Scholar]
- 11.This catalyst was prepared in situ by adding one equivalent of AgNTf2 to the commercially available chloride catalyst.
- 12.Tetraallyltin was found to be much more efficient than allyltributyltin.
- 13.Unni AK, Takenaka N, Yamamoto H, Rawal VH. J. Am. Chem. Soc. 2005;127:1336. doi: 10.1021/ja044076x. [DOI] [PubMed] [Google Scholar]
- 14.For a similar model see: Huang Y, Iwama T, Rawal VH. Org. Lett. 2002;4:1163. doi: 10.1021/ol0255716.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.