

Abstract
A systematic study of the structure–activity relationships (SAR) of 2b (OL-135), a potent inhibitor of fatty acid amide hydrolase (FAAH), is detailed targeting the C2 acyl side chain. A series of aryl replacements or substituents for the terminal phenyl group provided effective inhibitors (e.g., 5c, aryl = 1-napthyl, Ki = 2.6 nM) with 5hh (aryl = 3-Cl-Ph, Ki = 900 pM) being 5-fold more potent than 2b. Conformationally-restricted C2 side chains were examined and many provided exceptionally potent inhibitors of which 11j (ethylbiphenyl side chain) was established to be a 750 pM inhibitor. A systematic series of heteroatoms (O, NMe, S), electron-withdrawing groups (SO, SO2), and amides positioned within and hydroxyl substitutions on the linking side chain were investigated which typically led to a loss in potency. The most tolerant positions provided effective inhibitors (12p, 6-position S, Ki = 3 nM or 13d, 2-position OH, Ki = 8 nM) comparable in potency to 2b. Proteomic-wide screening of selected inhibitors from the systematic series of >100 candidates prepared revealed that they are selective for FAAH over all other mammalian serine proteases.
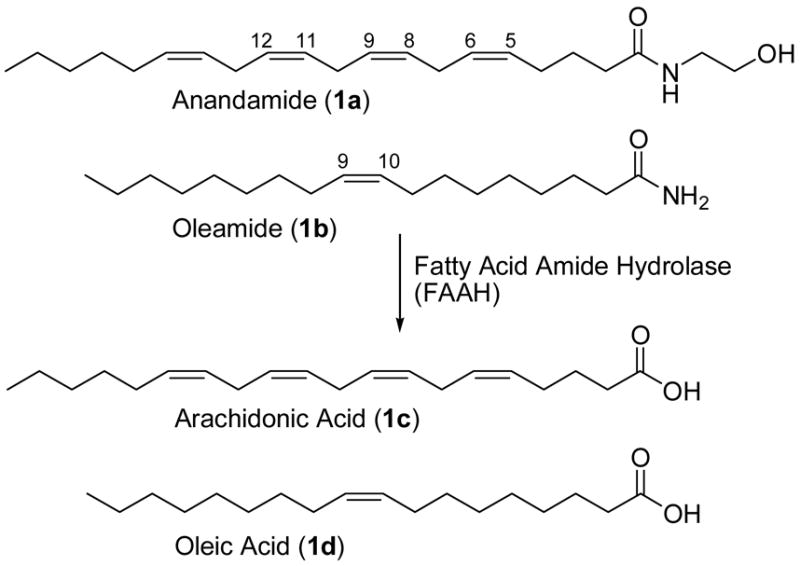
The enzyme fatty acid amide hydrolase (FAAH) is the primary catabolic regulator of several bioactive lipid amides in vivo, including anandamide (1a) and oleamide (1b).1–4 The central nervous system distribution of FAAH suggests that it degrades neuromodulating fatty acid amides at their sites of action and is intimately involved in their regulation.5 Fatty acid amide hydrolase is currently the only characterized mammalian enzyme that is in the amidase signature family bearing an unusual catalytic Ser-Ser-Lys triad.1,4,6–8 Recently, the crystal structure of FAAH cocrystallized with an irreversibly-bound arachidonyl fluorophosphonate confirmed its unusual catalytic triad and provided structural details of this enzyme.1
Both anandamide (1a)9 and oleamide (1b)10–12 have emerged as prototypical members of the class of bioactive lipid amides13,14 that serve as chemical messengers (Figure 1). Anandamide (1a), the most recognized member of the endogenous fatty acid ethanolamides,15 binds and activates both the central type-1 (CB1) and peripheral type-2 (CB2) cannabinoid receptors. Anandamide (1a), and members of the cannabinoid family,16 have been implicated in the modulation of nociception,17–19 feeding,20,21 emesis, anxiety,22 cell proliferation,23,24 inflammation,25 memory26 and neuroprotection after brain injury.27 Thus, the cannabinoids have clinical relevance for analgesia, anxiety, epilepsy, cachexia, cancer, and Alzheimer’s disease as well as other neurodegenerative diseases.28–30
Figure 1.
Substrates of fatty acid amide hydrolase (FAAH).
Oleamide (1b) was found to accumulate in the cerebrospinal fluid of animals under conditions of sleep deprivation and to induce physiological sleep in a dose dependent manner.10,12 It modulates serotonergic systems31–33 and GABAergic transmission,34,35 decreases body temperature and locomotor activity,36 and blocks glial gap junction cell-cell communication.37,38 The dual inhibition of presynaptic Na+ channels and postsynaptic GABAA receptors suggests oleamide (1b) may possess a mode of action common to drugs that are widely used for the treatment of anxiety, sleep disorders, and epilepsy and that it represents an endogenous ligand for such depressant drug sites in the mammalian brain. Oleamide (1b) decreases body temperature and locomotor activity,36 and exhibits the characteristic in vivo analgesic and cannabinoid behavorial effects of anandamide,31,39 albeit without apparent cannabinoid receptor activation. It has been suggested that the cannabinoid behavorial effects of oleamide (1b) may be mediated through an as yet unknown distinct pharmacological target.34 Because oleamide (1b) may play an important role in sleep, it may provide opportunities for the development of sleep aids that induce physiological sleep lacking the side effects of the sedative-hypnotics (e.g., benzodiazepene class), which include rebound insomnia, anterograde amnesia and suicide abuse potential.
The pharmacological actions of anandamide (1a) and oleamide (1b) are terminated by FAAH (Figure 1).1–4 Studies with FAAH knockout mice have not only shown that FAAH is a key regulator of fatty acid amide signaling in vivo, but that the animals exhibit a significantly augmented behavioral response (e.g., increased analgesia, hypomotility, catalepsy) to administered anandamide (1a) and oleamide (1b), that correlated with a CB1-dependent analgesic phenotype.40–43 As such, FAAH has emerged as an interesting new therapeutic target for a range of clinical disorders.16,44
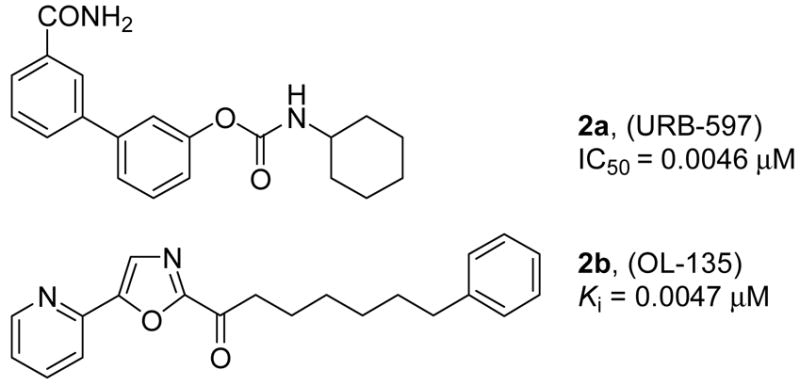
Due to the potentially exciting therapeutic potential of inhibiting FAAH, there has been increasing interest in the development of potent inhibitors (Figure 2).22,45–60 These include the discovery that the endogenous sleep-inducing molecule 2-octyl α-bromoacetoacetate is an effective FAAH inhibitor,55 a series of reversible inhibitors bearing an electrophilic ketone46,54,56 (e.g., trifluoromethyl ketone-based) that have not proven selective for FAAH over other mammalian serine hydrolases61 and a set of irreversible inhibitors48–50,52,53 (e.g., fluorophosphonates and sulphonyl flourides). Recently, two classes of inhibitors have been disclosed that provide significant opportunities for the development of an inhibitor with therapeutic potential. One class is the aryl carbamates (e.g., 2a; Figure 2) that acylate an active site catalytic serine and which were shown to exhibit anxiolytic activity and induce analgesia.22,57–59,62 However, the selectivity of such aryl carbamate inhibitors is low and recent studies illustrate that either no or minimal selectivity is achieved (e.g., other targets of 2a are carboxylesterase 6 and triacylglyceride hydrolase).63–65 A second class is the α-ketoheterocycle-based inhibitors of which some are extraordinarily potent (e.g., 2b; Figure 2).45,47,51,61,66 These competitive inhibitors bind to FAAH via reversible hemiketal formation with an active site serine, and are not only potent and extraordinarily selective for FAAH versus other mammalian serine hydrolases,45,61 but many are efficacious in vivo and promote analgesia.64,67
Figure 2.
FAAH Inhibitors.
From these latter studies, 2b has emerged as an advanced lead for further study.45,51,61,64,66 It has been shown that 2b is a potent and selective FAAH inhibitor that induces analgesia by raising endogenous anandamide levels,64 is ≥300-fold selective for FAAH over any other serine hydrolase,45 lacks significant offsite target activity when surveyed against a panel of receptors and enzymes (Cerep assay profiling), and does not significantly inhibit common P450 metabolism enzymes (3A4, 2C9, 2D6) or the human ether-a-go-go related gene (hERG). Consequently, we embarked on a more extensive study of the structure–activity relationships (SAR) of 2b. We recently reported a detailed study targeting the 5-position of the central oxazole of 2b (e.g., aryl and non-aromatic substituents) and disclosed a series of extraordinarily potent (as low as 400 pM) and selective FAAH inhibitors.68,69 Herein we report the synthesis and evaluation of a systematic series of α-keto oxazole inhibitors that extensively explores the C2 acyl side chain of 2b along with results of the proteome-wide selectivity screening of the candidate inhibitors.70
Chemistry
The majority of the candidate inhibitors were prepared by one of two methods (Scheme 1). Sonogashira coupling (Method A) of 3a45 or 3b45 with a series of aryl iodides afforded inhibitors 4e, 4f, 4pp, 4qq, 4o–4s, 4rr, 4ss, 4x–4cc, 4gg–4jj.71 Hydrogenation of the alkyne provided inhibitors 5e, 5f, 5o–5s, 5pp, 5qq, 5x–5cc, 5gg–5jj. Alternatively, direct acid chloride acylation (Method B) of a Zn/Cu-metalated 5-(pyridin-2-yl)oxazole72 (6) following the protocol of Anderson et al.73 yielded inhibitors 5a–5d, 5g–5l, 5t–5v, 5nn, 5oo, 11a–11j, 12e, 12g, 12i, 12k, 12n, 12p.
Scheme 1.

Scheme 2 summarizes the synthesis of the inhibitors that were not prepared by either Method A or B. Oxazole 6 was lithiated at C2, converted to its C2-stannane upon treatment with Bu3SnCl and subsequently treated with the corresponding acid chloride to afford 12a and 12b.74,75 Compound 12f was prepared by Michael addition of N-methyl-3-phenylpropylamine to enone 7. Treatment of 8 with TiCl4 followed by N-methyl-3-phenylethylamine provided 12h.76,77 Compounds 9j and 9o were transformed to a series of amino ketones upon treatment with the corresponding amine in the presence of K2CO3 in 2-butanone or by treatment with the neat amine. Esters 14a–14c were readily converted to their corresponding amides 13a–13c. α-Hydroxylation of OL-135 (2b) with the Davis reagent78 afforded 7 13d. Enolization of 10 with KHMDS and subsequent treatment with 5-phenylpentanal yielded 13e. Inhibitors 5kk–5mm, 12c, 12d, 121, 12m, 12q and 12r were prepared by oxidation (m-CPBA) of the corresponding sulfides 5t–v, 12b, 12k, and 12p.
Scheme 2.

Enzyme Assay
Enzyme assays were performed at 20–23 °C with purified recombinant rat FAAH expressed in E. coli79 (unless indicated otherwise) or with solubilized COS-7 membrane extracts from cells transiently transfected with human FAAH cDNA3 (where specifically indicated) in a buffer of 125 mM Tris/1 mM EDTA/0.2% glycerol/0.02% Triton X-100/0.4 mM Hepes, pH 9.0 buffer.55 The initial rates of hydrolysis (≤10–20% reaction) were monitored using enzyme concentrations (typically 1 nM) at least three times below the measured Ki by following the breakdown of 14C-oleamide and Ki’s (standard deviations are provided in Supporting Information tables) were established as described (Dixon plot).47 Lineweaver–Burk analysis previously established reversible, competitive inhibition for 2b and related inhibitors.45
Results and Discussion
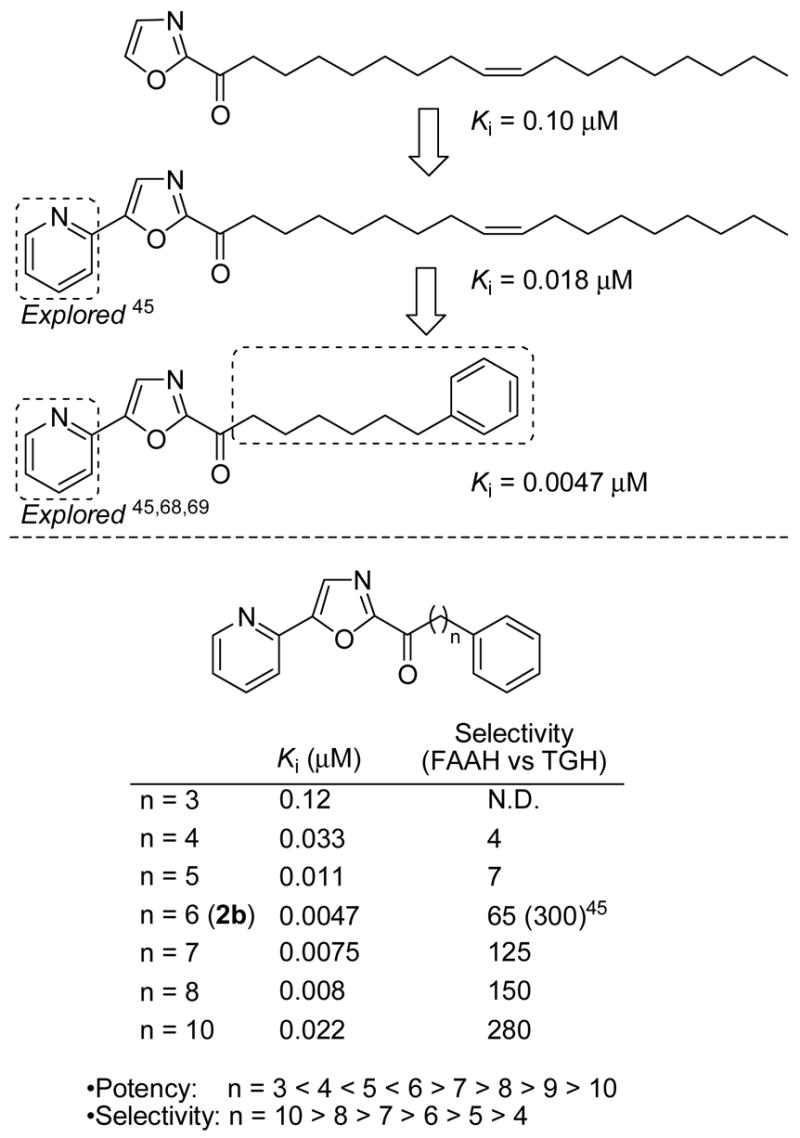
In preceding studies,45 the length of the linker chain and the incorporation of a terminal phenyl were found to impact and improve inhibitor potency (Figure 3). The linker chain length on compounds related to 2b exhibited a well-defined parabolic relationship with an optimal length of six-carbons terminating in a phenyl group constituting an overall length of C11 including the keto group. Slight perturbations on this structure were well tolerated although shortening the chain length generally led to reduced FAAH selectivity as well as potency, whereas lengthening the connecting chain improved selectivity, but with diminished FAAH potency. The studies herein explore the impact of further altering this C2 acyl side chain of 2b.
Figure 3.
Preceding studies of the C2 side chain.
Substitution of C2 Side Chain Terminal Phenyl Group
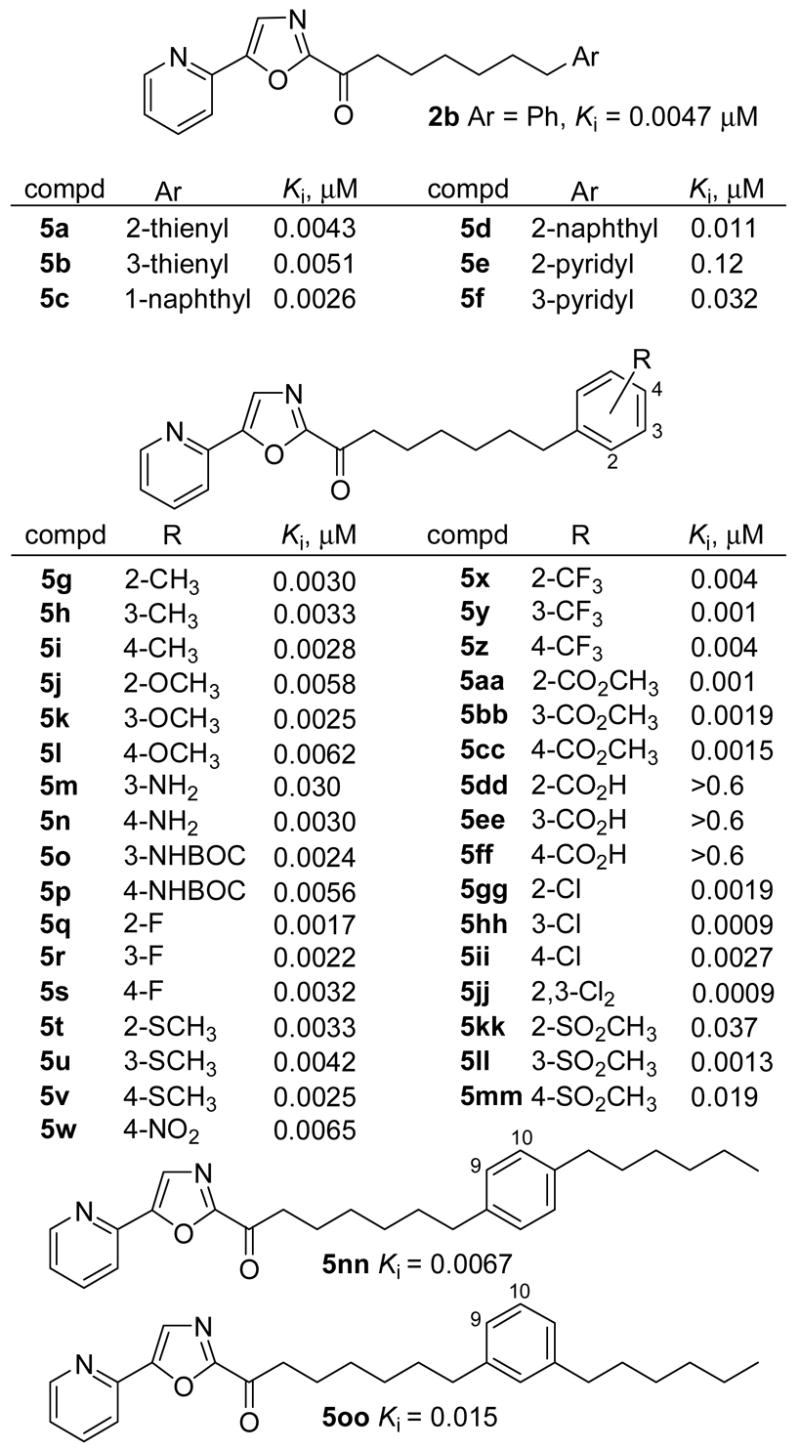
A systematic series of aryl replacements and phenyl substitutions for the terminal phenyl group of 2b was examined (Figure 4). Consistent with expectations based on a modeled FAAH active site analysis surrounding the bound phenyl ring of 2b,45 which corresponds to the site of π-interactions with the substrates as well as a bend in their bound conformations (Δ8,9/Δ11,12 double bonds of 1a, Δ9,10 double bond of 1b), almost all aryl replacement derivatives (5a–5f) proved to be effective inhibitors. Both thiophene replacements 5a and 5b were indistinguishable from 2b, the 1-naphthyl substitution 5c was roughly 2-fold more potent, and the 2-naphthyl derivative 5d was roughly 2-fold less potent. Incorporation of a more polar pyridine with 5e and 5f led to 7–25 fold reductions in the Ki with the 2-pyridiyl replacement being the most detrimental to inhibitory potency.
Figure 4.
Substitution of the C2 side chain terminal phenyl group.
Substitution of the terminal phenyl ring of 2b was broadly tolerated and the complete range of ortho, meta, or para substituents that were examined provided effective inhibitors (5g–5oo; Figure 4). The only exceptions to this generalization were the carboxylic acid derivatives (5dd–ff) which are deprotonated under the assay conditions (pH 9) and fail to effectively bind in the hydrophobic active site. Typically, hydrophobic or electron-withdrawing substituents enhanced the binding affinity of the inhibitors more significantly than polar or electron-donating substituents. However, and with a couple of notable exceptions, each substituent enhanced binding affinity indicative of additional favorable binding contacts within the active site. Although this may not be surprising for the hydrophobic substituents (CH3, CF3, F, Cl, SCH3 ≥ OCH3, H), it is especially interesting that polar substituents (CO2CH3, NO2, SO2CH3, NH2) can be tolerated in this hydrophobic pocket and that some even enhance inhibitory potency. This appears to be especially true of the m-position where even the methylsulfone 5ll produced an inhibitor more potent than 2b, whereas the corresponding o- and p-methylsulfone derivatives (5kk and 5mm, respectively) were ≥10-fold less effective. The potency of such derivatives typically ranged from 5–0.9 nM (Ki), displayed a variable and weak preference for the site of attachment, and the most potent members typically were the m-substituted derivatives. Significantly, 5hh (R = Cl) broke the nanomolar potency barrier providing a Ki of 900 pM and exceeding the activity of 2b by 5-fold. Accordingly, this region provides a rich area where substituents or modifications can be introduced to enhance inhibitor potency, impact features contributing to or improving in vivo properties, and substantially enhance selectivity.
Extending an alkyl m- or p-substituent such that the chain mimics the length of oleamide and the aryl ring placement mimics the position, conformation, and π-characteristics of the cis Δ9,10 double bond revealed that 5nn (p-substituent) matched the potency of 2b (but did not improve it), whereas 5oo (m-substituent) was less effective. These observations are analogous to the results of previous studies which additionally revealed a significant decrease in activity with an extended alkyl o-substituent.46
C2 Side Chain Conformational Constraints
Clear from a FAAH active site analysis and the inspection of the x-ray structure1 of a covalently bound arachidonyl fluorophosphonate, the lipid side chain can adopt a C4–C7 gauche versus extended conformation accommodating the Δ5,6 double bond of 1a and possesses sufficient active site space to accommodate bridges linking the C4/C7 gauche (cis) sites. In an effort to remove the rotatable side chain bonds in 2b potentially improving in vivo absorption characteristics, a series of diaryl containing side chains were examined which constrain the inhibitors in this gauche (cis) conformation mimicking the conformation and π-characteristics of the arachidonyl Δ5,6 double bond (Figure 5). A second terminal aryl group was incorporated to overlay with the arachidonyl Δ11,12 double bond and mimic the terminal phenyl group of 2b. Consistent with this analysis, 11a proved to be among one of the most potent FAAH inhibitors disclosed herein surpassing the activity of 2b. The isomer 11c was nearly 10-fold less potent, and incorporation of a heteroatom (O) into the side chain β to the carbonyl (11b and 11d) led to dramatic losses in potency despite its inductive electron-withdrawing character that might be expected to enhance the electrophilic character of the carbonyl. A survey of related compounds (11e–11i, Ki = 1–3 nM) revealed a wide tolerance for the functionality linking the two aryl rings and that its removal with ethylbiaryl 11j provided an exceptionally potent FAAH inhibitor (Ki = 750 pM).
Figure 5.
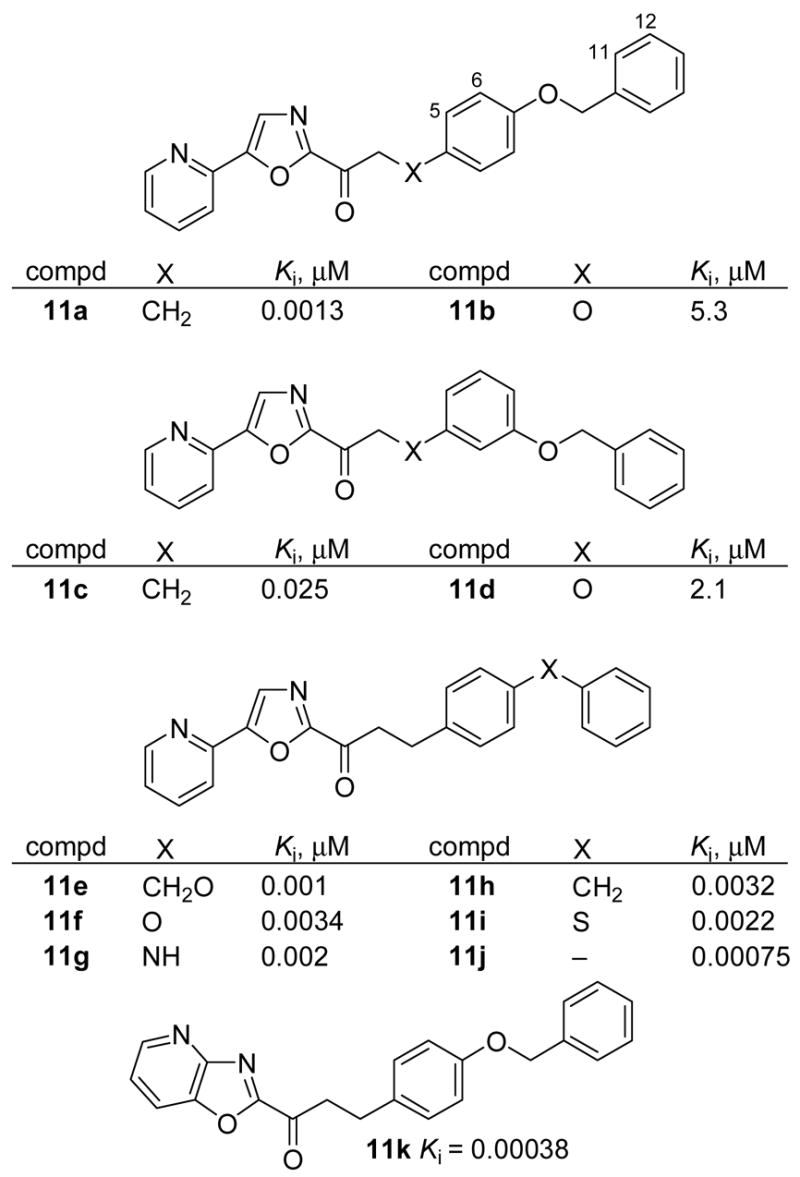
Conformationally restricted C2 side chain inhibitors.
In a previous study, the α-keto-5-(2-pyridyl)oxazol-2-yls were found to be roughly 5–25 fold less potent than their corresponding oxazolopyridines.47 Accordingly 11k (Ki = 380 pM) was also prepared and found to be roughly 5-fold more potent than 11a.
Finally, the alkyne precursors 4 to the series 5 inhibitors prepared by the Shonogashira coupling (Method A) were also examined for FAAH inhibition and the results are summarized in Figure 6. It was observed that there was a 2–20-fold loss in activity with the alkynes compared to their corresponding alkane derivatives (Figure 4) suggesting that this restriction places the terminal aryl ring in a less favorable area in the FAAH active site.
Figure 6.
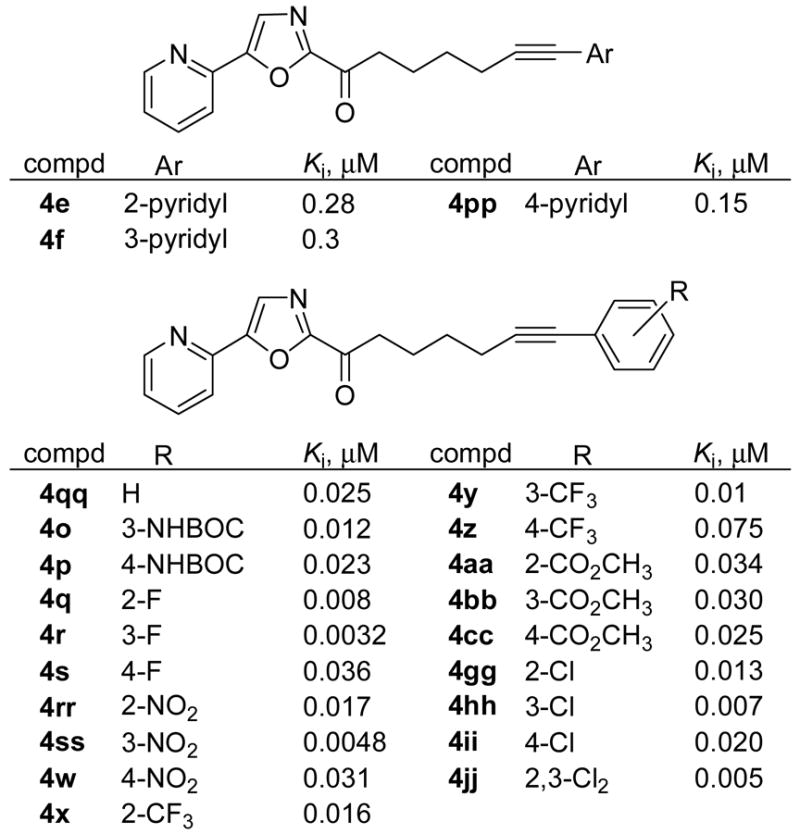
Linker alkynes.
Substitution along the side chain
A systematic series of heteroatoms and electron-withdrawing substituents positioned within the linking side chain of 2b was also investigated (Figure 7). Surprisingly, but consistent with observations made with 11b and 11d (Figure 5), placing a heteroatom or electron-withdrawing substitutent at the 2-position of the side chain (12a–12d) β to the electrophilic carbonyl resulted in a dramatic loss in potency compared to 2b despite their inductive electron-withdrawing character that would be expected to enhance the electrophilic character of the carbonyl. Only the least electronegative atom of the series (X = S, 12b) matched the potency of 2b and the most electronegative functionality (X = SO2, 12d) resulted in a 4000-fold loss in potency. A rough trend in the Ki is observed as heteroatoms move along the chain where heteroatoms at each end of the chain (2- and 6-positions) are better tolerated than in the middle (3–5 positions). At each location, the substitutions exhibited a well-defined trend of CH2 ≥ S > O > NMe > SO >SO2, albeit in a series that is not complete at each position, clearly reflecting the hydrophobic character of this region of FAAH active site. Thus, introduction of a sulfur atom provided inhibitors that were comparable (12b, 2-position; 12p, 6-position) or only slightly less potent (12k, 5-position) than 2b, whereas introduction of an oxygen (>10-fold), NMe (50 to 100-fold), SO (200 to 500-fold), and SO2 (400 to 1000-fold) led to progressive and substantial losses in binding affinity. At the most tolerant position (position 6), the magnitude of these effects for sulfur, oxygen, and NMe are dampened with each providing effective inhibitors.
Figure 7.
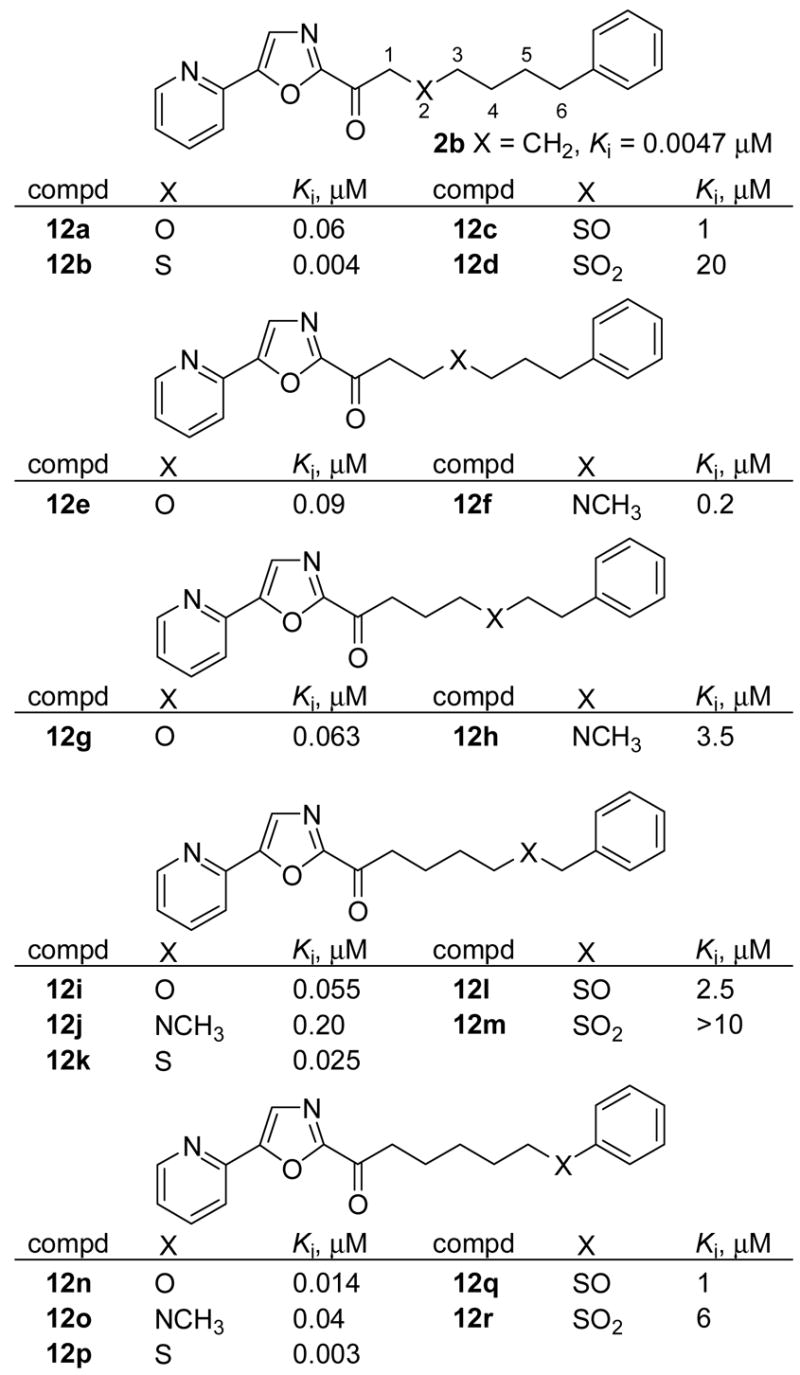
Incorporation of heteroatoms within the side chain.
A series of amides within the linking chain and hydroxyl substitutions on the chain was also explored (Figure 8). Amide placement in the side chain led to a dramatic loss in inhibitory potency (≥ 1000-fold). Consistent with expectations, 13a, but not 13b or 13c, exists as the stable N-acyl hemiaminal and this is reflected in its inability to inhibit FAAH. Consistent with the previous series of inhibitors (Figure 7), a well-defined trend in Ki is observed as the hydroxyl substitution moves along the side chain where the hydroxyl group at each end of the side chain was better tolerated than those in the middle of the side chain. Within this series, only 13f (50%, CDCl3) and 13g (50%, CDCl3) exist in equilibrium with their internal hemiketal. Thus, even though 13f is 100-fold less potent than 2b, it may prove useful to examine in vivo where the electrophilic carbonyl would potentially be less prone to metabolic reduction due to the reversible hemiketal formation.
Figure 8.
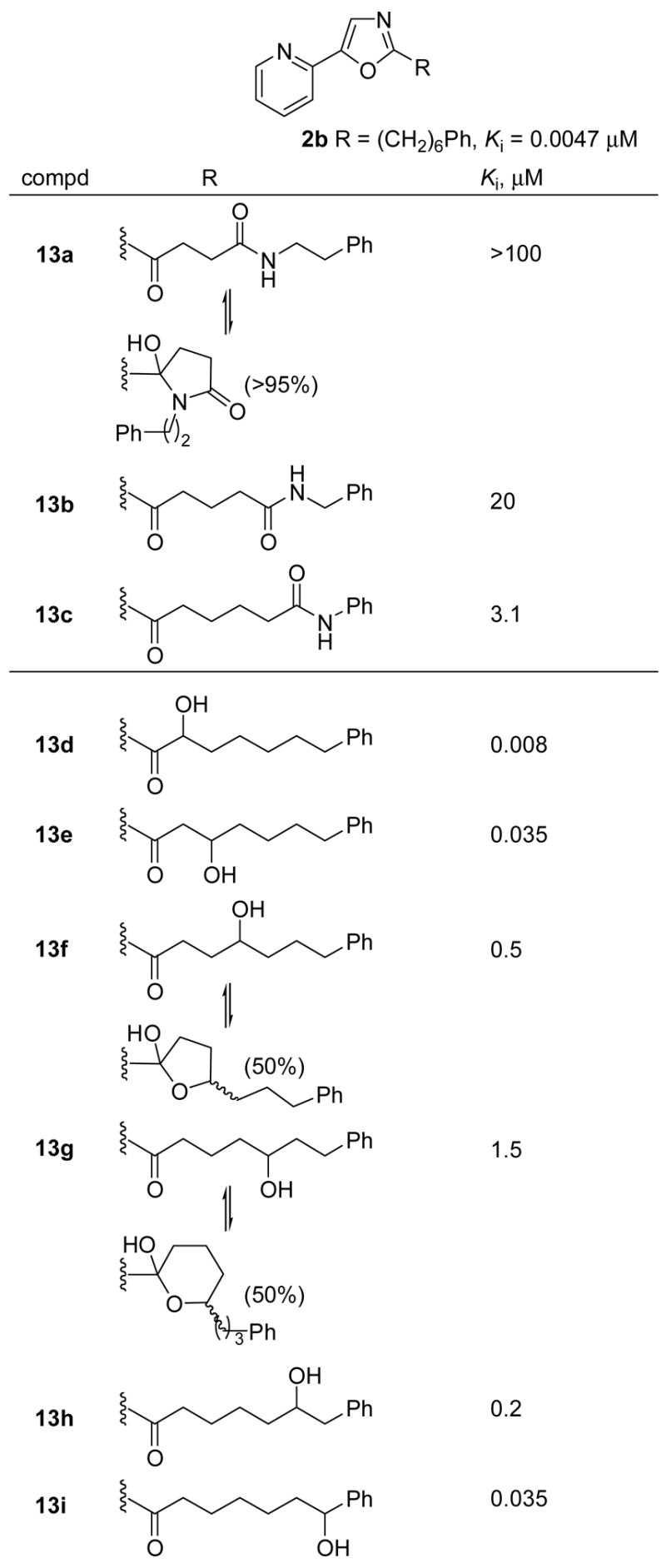
Hydroxyl and amide substitution.
Several additional side chain modifications were examined and represent intermediates or byproducts derived from the synthesis of the preceding candidate inhibitors. The results of their examination are summarized in Figure 9 and highlight several features. Shortening of the side chain and removal of the phenyl group of 2b with a small series of methyl esters (14a–14c) led to a significant progressive decrease in potency. The cyclopropyl and cyclopentyl derivatives 14d and 14e lacking the extended chain and phenyl group similarly resulted in a loss of activity. Interestingly, and in contrast, the simple α-chloroketone 14f was still a submicromolar inhibitor of FAAH although it lacked nearly all of the C2 side chain. Presumably this reflects the inhibitors increased electrophilic carbonyl reactivity increasing its potency ≥100-fold over the inactive methyl ketone45 (Ki = >100 μM). Placement of a ketone β to the electrophilic carbonyl (14g) led to a complete loss of inhibitory potency compared to 2b. In this instance, the electrophilic C2 carbonyl of 14g is enolized (>95%, CDCl3) and unreactive toward nucleophilic attack.
Figure 9.
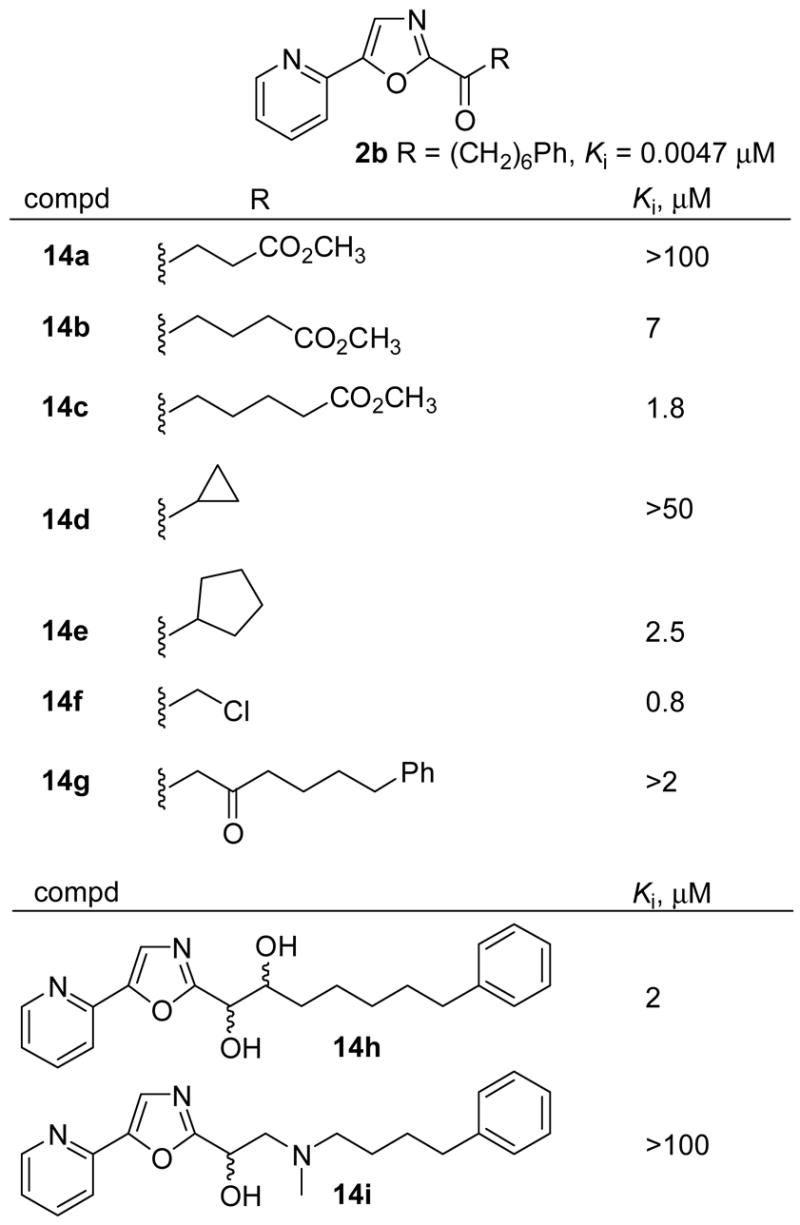
Effect of additional side chain modifications.
Finally two alcohol derivatives were examined and consistent with previous observations,45 14h and 14i both resulted in a substantial loss in inhibitory potency with 14h exhibiting a 250-fold loss in activity relative to its corresponding ketone 13d.
Inhibition of Recombinant Human FAAH
Rat and human FAAH are very homologous (84% sequence identity), exhibit near identical substrate selectivities and inhibitor sensitivities in studies disclosed to date,47 and embody an identical amidase signature sequence suggesting the observations made with rat FAAH would be analogous to those made with the human enzyme. Consequently, key inhibitors in the series were examined against the human enzyme and consistent with previous observations45,69 were found to exhibit the same relative and absolute potencies (Figure 10).
Figure 10.
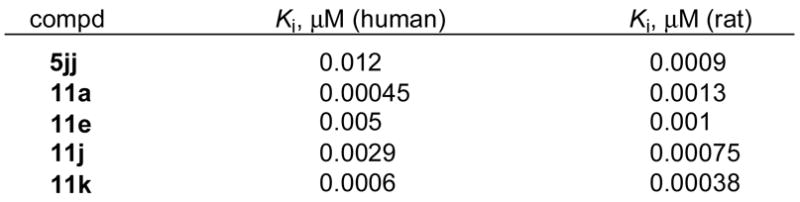
Inhibition of Recombinant Human Fatty Acid Amide Hydrolase.
Selectivity Screening
Early assessments of α-ketoheterocycle inhibitors of FAAH against possible competitive enzymes (e.g., phospholipase A2, ceramidase) revealed no inhibition. Consequently a method for proteomic-wide screening capable of globally profiling all mammalian serine hydrolases was developed70 and studies have shown that the α-ketoheterocycle class of inhibitors generally are exquisitely selective for FAAH.45,61,64 However, two enzymes did emerge as potential competitive targets: triacylglycerol hydrolase (TGH) and an uncharacterized membrane-associated hydrolase that lacks known substrates or function (KIAA1363). In this screen, IC50 values are typically higher than the measured Ki values, but the relative potency, the magnitude of binding affinity differences and the rank order binding determined in the assay parallels that established by standard substrate assays.
Summarized in Figure 11 are the results of the selectivity screening of selected candidate inhibitors. In general, the inhibitors, like 2b, were very selective for FAAH over TGH and KIAA1361. The pyridyl replacements (5e, 5f) of the terminal phenyl group of 2b proved very selective for FAAH over KIAA1363, but only moderately selective for FAAH over TGH. Substitution on the terminal phenyl ring of 2b also provided selective inhibitors and this was relatively independent of the substitution position (o-, m-, or p-) and whether it was electron-donating or electron-withdrawing (5j–5l vs. 5gg–5ii). Inhibitors that possess the side chain conformational constraints (11a, 11e–11j) generally were very selective for FAAH over KIAA1363 and typically 100- to 1000-fold selective for FAAH over TGH. Most notably, 11a, 11e, and 11j are the most selective inhibitors of the series surpassing 2b in both their FAAH potency and selectivity.
Figure 11.
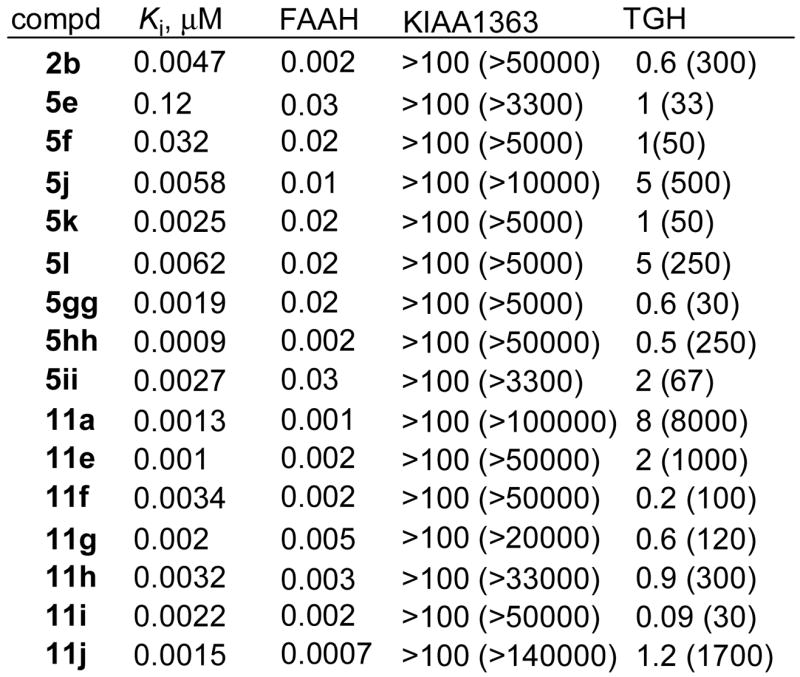
Selectivity screening; IC50 (selectivity). A full table of results is reported in Supporting Information.
Conclusion
In previous studies,45 the length of the C2 acyl side chain and the incorporation of a terminal phenyl group were found to impact and improve FAAH inhibitor potency leading to the discovery of 2b. To more thoroughly explore the C2 side chain of 2b, an extensive series of more than 100 derivatives was prepared and evaluated for FAAH inhibitory potency as well as FAAH selectivity versus competitive serine proteases (e.g., TGH, KIAA1363). Aryl replacements of the terminal phenyl group of 2b resulted in effective inhibitors of which many are indistinguishable from 2b and notably with 5c (aryl = 1-napthyl, Ki = 2.6 nM) being ca. 2-fold more potent than 2b. A large series of phenyl substituents also proved to be effective inhibitors with hydrophobic or electron-withdrawing meta substituents generally enhancing binding affinity to the greatest extent with 5hh (aryl = 3-Cl-Ph, Ki = 900 pM) being 5-fold more potent than 2b. Conformationally restricted C2 side chains of 2b were examined and many of these were found to be exceptionally potent of which 11j (ethylbiphenyl side chain) is a 750 pM inhibitor of FAAH. A systematic series of heteroatoms (O, NMe, S) and electron-withdrawing groups (SO, SO2) positioned within the linking side chain of 2b were investigated and, surprisingly, substitution β to the electrophilic carbonyl led to a dramatic loss in potency. The most tolerant position (position 6) provided effective inhibitors (12p, X = S, Ki = 3 nM) comparable to 2b. A series of amides within the linking chain and hydroxyl substitutions on the chain were also explored. Amide placement in the side chain led to a dramatic loss in inhibitory potency whereas hydroxyl substitution at positions 2 and 6 provided effective inhibitors (13a, 2-position OH, Ki = 8 nM). Just as importantly, proteome-wide selectivity screening of the candidate inhibitors showed extraordinary selectivity for FAAH over all other serine hydrolases and proteases. Most notably, inhibitors that possess the side chain conformational constraints (11a, 11e–11j) generally were very selective for FAAH over KIAA1363 and typically 100- to 1000-fold selective for FAAH over TGH. Finally and despite the lipophilic and non drug-like nature of the FAAH substrates, the lead structure 2b and the potent inhibitors disclosed herein including 5hh and 11j possess much more favorable drug-like characteristics.80
Experimental
1-Oxo-1-[5-(2-pyridyl)oxazol-2-yl]-7-(3-chlorophenyl)heptane (5hh)
A solution of hept-6-ynoic acid (1.90 g, 14.8 mmol) in anhydrous THF (90 mL) at −78 °C was treated with n-BuLi (2.3 M in hexanes, 14.5 mL, 33.3 mmol). After stirring for 2 min, TMSCl (5.8 mL, 46.0 mmol) was added. The reaction mixture was allowed to warm slowly to 25 °C and was stirred for 1 h. The reaction was quenched with the addition of aqueous 2 N HCl and extracted with CH2Cl2. The organic layer was dried over Na2SO4, filtered and concentrated. Column chromatography (SiO2, 4 × 6 cm, 20% EtOAc–hexanes) afforded 7-(trimethylsilyl)hept-6-ynoic acid (2.7 g, 13.6 mmol, 92%) as a white solid: 1H NMR (CDCl3, 500 MHz) δ 2.40 (t, 2H, J = 7.4 Hz), 2.24 (t, 2H, J = 7.3 Hz), 1.78–1.72 (m, 2H), 1.62–1.56 (m, 2H), 0.15 (s, 9H).
A solution of 5-(2-pyridyl)oxazole72 (600 mg, 4.11 mmol) in anhydrous THF (15 mL) at −78 °C was treated dropwise with a solution of n-BuLi (2.2 M in hexanes, 2.4 mL, 4.52 mmol,) under N2 and the resulting solution was stirred at −78 °C for 20 min. A solution of ZnCl2 (0.5 M in THF, 18 mL, 8.22 mmol,) was added, and the mixture was warmed to 0 °C. After stirring at 0 °C for 45 min, CuI (850 mg, 4.46 mmol) was added to the mixture. After the mixture was stirred at 0 °C for 10 min, a solution of 7- (trimethylsilyl)hept-6-ynoyl chloride (1.2 equiv; prepared from 7-(trimethylsilyl)hept-6-ynoic acid and oxalyl chloride) in anhydrous THF (9 mL) was added dropwise, and the mixture was stirred at 0 °C for an additional 1 h. The reaction mixture was diluted with a 1:1 mixture of hexanes and EtOAc (60 mL) and washed with 15% aqueous NH4OH (2 × 30 mL), water (30 mL) and saturated aqueous NaCl (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated. Column chromatography (SiO2, 4 × 6 cm, 30% EtOAc–hexanes) afforded 1-oxo-1-[5-(2-pyridyl)oxazol-2-yl]-7- (trimethylsilyl)hept-6-yne (3a, 875 mg, 2.68 mmol, 74%) as a tan oil: 1H NMR (CDCl3, 400 MHz) δ 8.68 (m, 1H), 7.89–7.86 (m, 2H), 7.82 (td, 1H, J = 7.6, 1.8 Hz), 7.34–7.31 (m, 1H), 3.15 (t, 2H, J = 7.3 Hz), 2.30 (t, 2H, J = 7.2 Hz), 1.94–1.86 (m, 2H), 1.68–1.60 (m, 2H), 0.14 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 187.9, 157.2, 153.2, 150.0, 146.1, 137.0, 126.8, 124.1, 120.3, 106.6, 84.8, 38.4, 27.9, 22.9, 19.6, 0.0; IR (film) νmax 2955, 2867, 2173, 1699, 1603, 1576, 1504, 1469, 1426, 1383, 1249, 1152, 1118, 1083, 1024, 929, 842, 784, 760 cm−1; ESI–TOF m/z 327.1530 (C18H22N2O2Si + H+ requires 327.1523).
A solution of 1-oxo-1-[5-(2-pyridyl)oxazol-2-yl]-7-(trimethylsilyl)hept-6-yne (3a, 570 mg, 1.75 mmol, 1 equiv) in anhydrous THF (6 mL) at 0 °C was treated with a solution of Bu4NF in THF (1 M, 2.1 mL, 2.1 mmol). After stirring for 35 min at 0 °C, the reaction mixture was quenched with H2O and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated. Column chromatography (SiO2, 2.5 × 3 cm, 30% EtOAc–hexanes) afforded 1-oxo-1-[5-(2- pyridyl)oxazol-2-yl]-hept-6-yne (3b, 340 mg, 1.36 mmol, 77%) as a tan solid: 1H NMR (CDCl3, 500 MHz) δ 8.68–8.66 (m, 1H), 7.89–7.86 (m, 2H), 7.82 (td, 1H, J = 7.6, 1.8 Hz), 7.34–7.31 (m, 1H), 3.15 (t, 2H, J = 7.3 Hz), 2.27 (td, 2H, J = 7.2, 2.7 Hz), 1.96 (t, 2H, J = 2.7 Hz), 1.94–1.88 (m, 2H), 1.68–1.62 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 187.9, 157.2, 153.2, 150.1, 146.2, 137.1, 126.8, 124.1, 120.3, 83.8, 68.7, 38.4, 27.7, 22.9, 18.2; IR (film) ν max 2938, 2867, 2115, 1698, 1603, 1575, 1505, 1470, 1426, 1385, 1283, 1245, 1127, 1086, 1024, 991, 962, 853, 785, 743 cm−1; ESI–TOF m/z 255.1135 (C15H14N2O2 + H+ requires 255.1128).
A solution of 1-chloro-3-iodobenzene (49 mg, 0.205 mmol) in anhydrous THF (0.5 mL) was treated with PdCl2(PPh3)2 (7 mg, 0.01 mmol). After stirring for 5 min at 25 °C, Et3N (0.2 mL, 0.603 mmol) and CuI (10 mg, 0.053 mmol) were added. The suspension was stirred for 35 min and 1-oxo-1-[5-(2- pyridyl)oxazol-2-yl]-hept-6-yne (3b, 30 mg, 0.067 mmol) was added. After stirring for 14 h at 25 °C, the reaction mixture was filtered through Celite and concentrated. PTLC (SiO2, 50% EtOAc–hexanes) afforded 1-oxo-1-[5-(2-pyridyl)oxazol-2-yl]-7-(3-chlorophenyl)hept-6-yne (4hh, 24 mg, 0.066 mmol, 56%) as a yellow solid: mp 50–51 °C; 1H NMR (CDCl3, 500 MHz) δ 8.68–8.66 (m, 1H), 7.89–7.86 (m, 2H), 7.82 (td, 1H, J = 7.7, 1.8 Hz), 7.38 (m, 1H), 7.34–7.31 (m, 1H), 7.27–7.18 (m, 3H), 3.20 (t, 2H, J = 7.4 Hz), 2.49 (t 2H, J = 7.0 Hz), 2.00–1.95 (m, 2H), 1.77–1.71 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 187.9, 157.2, 153.3, 150.1, 146.2, 137.1, 133.9, 131.4, 129.6, 129.3, 127.8, 126.8, 125.5, 124.1, 120.3, 90.9, 79.8, 38.5, 27.8, 23.1, 19.1; IR (film) νmax 3061, 2932, 2865, 2230, 1703, 1592, 1575, 1558, 1505, 1471, 1426, 1385, 1283, 1243, 1152, 1081, 1065, 1023, 990, 962, 930, 880, 784, 740, 683 cm−1; ESI–TOF m/z 365.1058 (C21H17ClN2O4 + H+ requires 365.1051).
A solution of the oxo-1-[5-(2-pyridyl)oxazol-2-yl]-7-(3-chlorophenyl)hept-6-yne (4hh, 15 mg, 0.041 mmol) in anhydrous THF (1 mL) was treated with a catalytic amount of Raney nickel (washed before use with THF). The reaction mixture was purged with H2 and stirred at 25 °C overnight. The suspension was filtered through Celite and concentrated. The crude product was dissolved in anhydrous CH2Cl2 (2 mL) and treated with Dess–Martin reagent (29 mg, 0.068 mmol). After stirring for 3 h at 25 °C, the reaction mixture was quenched with saturated aqueous Na2CO3 and saturated aqueous Na2S2O3. After stirring for 15 min, the mixture was extracted with CH2Cl2. The organic layer was dried over Na2SO4, filtered and concentrated. PTLC (SiO2, 40% EtOAc–hexanes) afforded the title compound (5hh, 10 mg, 0.027 mmol, 67%) as a white solid: mp 91–92 °C; 1H NMR (CDCl3, 600 MHz) δ 8.68–8.66 (m, 1H), 7.89–7.86 (m, 2H), 7.82 (td, 1H, J = 7.8, 1.4 Hz), 7.34–7.31 (m, 1H), 7.21–7.14 (m, 3H), 7.04 (d, 1H, J = 7.5 Hz), 3.11 (t, 2H, J = 7.4 Hz), 2.59 (t, 2H, J = 7.4 Hz), 1.81–1.76 (m, 2H), 1.65–1.60 (m, 2H), 1.46–1.36 (m, 4H); 13C NMR (CDCl3, 125 MHz) δ 188.4, 157.3, 153.2, 150.1, 146.3, 144.7, 137.1, 133.9, 129.5, 128.5, 126.8, 126.6, 124.1, 120.4, 39.0, 35.5, 31.0, 28.9, 28.8, 23.8; IR (film) ν max 2930, 2856, 1698, 1601, 1575, 1505, 1470, 1426, 1385, 1285, 1081, 1035, 990, 962, 935, 783, 741, 696 cm−1; ESI–TOF m/z 369.1363 (C21H21ClN2O2 + H+ requires 369.1364).
1-Oxo-1-[5-(2-pyridyl)oxazol-2-yl]-3-(4-(benzyloxy)phenyl)propane (11a)
4-Hydroxycinnamic acid (700 mg, 4.26 mmol) was dissolved in EtOAc (15 mL) and 10% Pd/C (51 mg, 0.479 mmol) was added. The reaction mixture was stirred under an atmosphere of H2 overnight at room temperature before it was filtered through Celite and concentrated in vacuo. No further purification was needed to yield 3-(4-hydroxyphenyl)propanoic acid (700 mg, 99%). A solution of 3-(4-hydroxyphenyl)propanoic acid (700 mg, 4.21 mmol) in anhydrous DMF (16 mL) at 0 °C was treated with a solution of 60% NaH (450 mg, 18.75 mmol) in DMF dropwise. The reaction mixture was stirred for 10 min before benzyl bromide (0.675 mL, 5.68 mmol) was added. The reaction mixture was stirred overnight at room temperature, quenched with aqueous 1 N HCl and extracted with EtOAc. The combined organic layers were washed with saturated aqueous NH4Cl, saturated aqueous NaCl and dried over Na2SO4. Column chromatography (SiO2, 4 × 9 cm, 20–40% EtOAc–hexanes gradient) afforded 3-(4- (benzyloxy)phenyl)propanoic acid (780 mg, 72%) as a white solid: 1H NMR (CDCl3, 500 MHz) δ 7.44 (d, 2H, J = 7.4 Hz), 7.40 (t, 2H, J = 7.4 Hz), 7.35–7.32 (m, 1H), 7.14 (d, 2H, J = 8.8 Hz), 6.92 (d, 2H, J = 8.4 Hz), 5.06 (s, 2H), 2.92 (t, 2H, J = 7.7 Hz), 2.66 (t, 2H, J = 7.7 Hz); 13C NMR (CDCl3, 125 MHz) δ 179.0, 157.3, 137.0, 132.5, 129.2, 128.5, 127.9, 127.4, 114.9, 70.0, 35.8, 29.7.
A solution of 5-(2-pyridyl)oxazole72 (6, 116 mg, 0.794 mmol) in anhydrous THF (4 mL) at −78 °C was treated dropwise with a solution of n-BuLi in hexanes (1.6 M, 0.64 mL, 0.953 mmol) under N2 and the resulting solution was stirred at −78 °C for 35 min. A solution of ZnCl2 in THF (0.5 M, 1.9 mL, 1.56 mmol) was added and the mixture was allowed to warm to 0 °C. After stirring at 0 °C for 45 min, CuI (160 mg, 0.840 mmol) was added. After the mixture was stirred at 0 °C for 15 min, a solution of 3- (4-hydroxyphenyl)propanoyl chloride (1.2 equiv; prepared from 3-(4-(benzyloxy)phenyl)propanoic acid and oxalyl chloride) in anhydrous THF (1.5 mL) was added dropwise, and the mixture was stirred for an additional 1 h. The reaction mixture was quenched with addition of saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was filtered through Celite, dried over anhydrous Na2SO4, filtered and evaporated to yield the crude product. Column chromatography (SiO2, 2.5 × 5 cm, 10–30% EtOAc–hexanes gradient) followed by PTLC (SiO2, 50% EtOAc–hexanes) afforded the title compound (11a, 33%) as a white solid: mp 99–100 °C 1H NMR (CDCl3, 400 MHz) δ 8.67 (app d, J = 4.4 Hz, 1H), 7.88 (s, 1H), 7.87–7.85 (m, 1H), 7.81 (td, 1H, J = 7.8, 1.8 Hz), 7.44 (d, 2H, J = 7.0 Hz), 7.39 (t, 2H, J = 7.5 Hz), 7.32 (t, 2H, J = 6.8 Hz), 7.19 (d, 2H, J = 8.5 Hz), 6.91 (d, 2H, J = 8.5 Hz), 5.04 (s, 2H), 3.44 (t, 2H, J = 7.4 Hz), 3.06 (t, 2H, J = 7.4 Hz); 13C NMR (CDCl3, 100 MHz) δ 187.4, 157.2 (2C), 153.2, 150.1, 146.2, 137.1, 137.0, 132.7, 129.4, 128.5, 127.9, 127.4, 126.9, 124.2, 120.4, 114.9, 70.0, 40.9, 28.9; IR (film) νmax 3097, 2919, 1693, 1602, 1582, 1514, 1470, 1427, 1382, 1253, 1177, 1042, 963, 938, 785, 741, 697 cm−1; ESI–TOF m/z 385.1549 (C24H20N2O3 + H+ requires 385.1547).
FAAH Inhibition
14C-labeled oleamide was prepared from 14C-labeled oleic acid as described.12 The truncated rat FAAH (rFAAH) was expressed in E. coli and purified as described.79 The purified recombinant rFAAH was used in the inhibition assays unless otherwise indicated. The full-length human FAAH (hFAAH) was expressed in COS-7 cells as described,3 and the lysate of hFAAH-transfected COS-7 cells was used in the inhibition assays where explicitly indicated.
The inhibition assays were performed as described.12 In brief, the enzyme reaction was initiated by mixing 1 nM of rFAAH (800, 500, or 200 pM rFAAH for inhibitors with Ki ≤ 1–2 nM) with 10 μM of 14C-labeled oleamide in 500 μL of reaction buffer (125 mM TrisCl, 1 mM EDTA, 0.2% glycerol, 0.02% Triton X-100, 0.4 mM Hepes, pH 9.0) at room temperature in the presence of three different concentrations of inhibitor. The enzyme reaction was terminated by transferring 20 μL of the reaction mixture to 500 μL of 0.1 N HCl at three different time points. The 14C-labeled oleamide (substrate) and oleic acid (product) were extracted with EtOAc and analyzed by TLC as detailed.12 The Ki of the inhibitor was calculated using a Dixon plot as described (standard deviations are provided in the Supporting Information tables).47 Lineweaver-Burk analysis was performed as described confirming competitive, reversible inhibition.45
Selectivity Screening
The selectivity screening was conducted as detailed.70
Supplementary Material
Full experimental details and characterization, FAAH assay measurement errors, and purities (HPLC analysis) of the FAAH inhibitors disclosed herein. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (Grant DA15648, D.L.B.; Grants DA017259 and DA015197, B.F.C.) and the Skaggs Institute for Chemical Biology, and the postdoctoral fellowship support for F.A.R. (American Cancer Society).
Abbreviations
- FAAH
fatty acid amide hydrolase
- TGH
triacylglycerol hydrolase
References
- 1.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural Adaptations in a Membrane Enzyme that Terminates Endocannabinoid Signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 2.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular Characterization of an Enzyme that Degrades Neuromodulatory Fatty Acid Amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 3.Giang DK, Cravatt BF. Molecular Characterization of Human and Mouse Fatty Acid Amide Hydrolases. Proc Natl Acad Sci US A. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patricelli MP, Cravatt BF. Proteins Regulating the Biosynthesis and Inactivation of Neuromodulatory Fatty Acid Amides. Vit Hormones. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 5.Egertova M, Cravatt BF, Elphick MR. Comparative Analysis of Fatty Acid Amide Hydrolase and CB1 Cannabinoid Receptor Expression in the Mouse Brain: Evidence of a Widespread Role for Fatty Acid Amide Hydrolase in Regulation of Endocannabinoid Signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 6.Patricelli MP, Cravatt BF. Fatty Acid Amide Hydrolase Competitively Degrades Bioactive Amides and Esters Through a Nonconventional Catalytic Mechanism. Biochemistry. 1999;38:14125–14130. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]
- 7.Patricelli MP, Cravatt BF. Clarifying the Catalytic Roles of Conserved Residues in the Amidase Signature Family. J Biol Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]
- 8.Patricelli MP, Lovato MA, Cravatt BF. Chemical and Mutagenic Investigations of Fatty Acid Amide Hydrolase: Evidence for a Family of Serine Hydrolases with Distinct Catalytic Properties. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 9.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and Structure of a Brain Constituent that Binds to the Cannabinoid Receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 10.Boger DL, Henriksen SJ, Cravatt BF. Oleamide: An Endogenous Sleep-Inducing Lipid and Prototypical Member of a New Class of Lipid Signalling Molecules. Curr Pharm Des. 1998;4:303–314. [PubMed] [Google Scholar]
- 11.Cravatt BF, Lerner RA, Boger DL. Structure Determination of an Endogenous Sleep-Inducing Lipid, cis-9-Octadecenamide (Oleamide): A Synthetic Approach to the Chemical Analysis of Trace Quantitites of a Natural Product. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 12.Cravatt BF, Prospero-Garcia O, Suizdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Chemical Characterization of a Family of Brain Lipids that Induce Sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 13.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Fatty Acid Amide Hydrolase Substrate Specificity. Bioorg Med Chem Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 14.Lang W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, Abouzid K, Meng Z, Biegel D, Makriyannis A. Substrate Specificity and Stereoselectivity of Rat Brain Microsomal Anandamide Amidohydrolase. J Med Chem. 1999;42:896–902. doi: 10.1021/jm980461j. [DOI] [PubMed] [Google Scholar]
- 15.Schmid HHO, Schmid PC, Natarajan V. N-Acylated Glycerophospholipids and Their Derivatives. Prog Lipid Res. 1990;29:1–43. doi: 10.1016/0163-7827(90)90004-5. [DOI] [PubMed] [Google Scholar]
- 16.Lambert DM, Fowler CJ. The Endocannabinoid System: Drug Targets, Lead Compounds, and Potential Therapeutic Applications. J Med Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 17.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of Pain Initiation by Endogenous Cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 18.Cravatt BF, Lichtman AH. The Endogenous Cannabinoid System and Its Role in Nociceptive Behavior. J Neurobiol. 2004;61:149–160. doi: 10.1002/neu.20080. [DOI] [PubMed] [Google Scholar]
- 19.Walker JM, Huang SM, Stragman NM, Tsou K, Sanudo-Pena MC. Pain Modulation by Release of the Endogenous Cannabinoid Anamdamide. Proc Natl Acad Sci US A. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del Arco I, Cippitelli A, Nava F, Piomelli D, Rodríguez de Fonseca F. A Peripheral Mechanism for CB1 Cannabinoid Receptor Dependent Modulation of Feeding. J Neurosci. 2002;22:9612–9617. doi: 10.1523/JNEUROSCI.22-21-09612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams CM, Kirkham TC. Observational Analysis of Feeding Induced by Δ-THC and Anandamide. Physiol Behav. 2002;76:241–250. doi: 10.1016/s0031-9384(02)00725-4. [DOI] [PubMed] [Google Scholar]
- 22.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of Anxiety Through Blockade of Anandamide Hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 23.Melck D, Rueda D, Galve-Roberh I, De Petrocellis L, Guzmán M, Di MV. Involvement of the cAMP/Protein Kinase A Pathway and of Mitogen-Activated Protein Kinase in the Anti-Proliferative Effects of Anandamide in Human Breast Cancer Cells. FEBS Lett. 1999;463:235–240. doi: 10.1016/s0014-5793(99)01639-7. [DOI] [PubMed] [Google Scholar]
- 24.Yamaji K, Sarker KP, Kawahara K, Iino S, Yamakuchi M, Abeyama K, Hashiguchi T, Maruyama I. Anandamide Induces Apoptosis in Human Endothelial Cells: Its Regulation System and Clinical Implications. Thromb Haemostasis. 2003;89:875–884. [PubMed] [Google Scholar]
- 25.Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B. The Endogenous Cannabinoid System Protects Against Colonic Inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mallet PE, Beninger RJ. The Cannabinoid CB1 Receptor Antagonist SR141716A Attenuates the Memory Impairment Produced by Δ9-Tetrahydrocannabinol or Anandamide. Psychopharmacology. 1998;140:11–19. doi: 10.1007/s002130050733. [DOI] [PubMed] [Google Scholar]
- 27.Panikashvili D, Simeonidou C, Ben Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An Endogenous Cannabinoid (2-AG) is Neuroprotective after Brain Injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 28.Axelrod J, Felder CC. Cannabinoid Receptors and Their Endogenous Agonist, Anandamide. Neurochem Res. 1998;23:575–581. doi: 10.1023/a:1022418217479. [DOI] [PubMed] [Google Scholar]
- 29.Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Martin BR. Cannabimimetic Fatty Acid Derivatives: The Anandamide Family and Other “Endocannabinoids” . Curr Med Chem. 1999;6:721–744. [PubMed] [Google Scholar]
- 30.Martin BR, Mechoulam R, Razdan RK. Discovery and Characterization of Endogenous Cannabinoids. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- 31.Cheer JF, Cadogan AK, Marsden CA, Fone KCF, Kendall DA. Modification of 5-HT2 Receptor Mediated Behaviour in the Rat by Oleamide and the Role of Cannabinoid Receptors. Neuropharmacology. 1999;38:533–541. doi: 10.1016/s0028-3908(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 32.Thomas EA, Cravatt BF, Sutcliffe JG. The Endogenous Lipid Oleamide Activates Serotonin 5-HT7 Neurons in Mouse Thalamus and Hypothalamus. J Neurochem. 1999;72:2370–2378. doi: 10.1046/j.1471-4159.1999.0722370.x. [DOI] [PubMed] [Google Scholar]
- 33.Boger DL, Patterson JE, Jin Q. Structural Requirements for 5-HT2A and 5-HT1A Receptor Potentiation by the Biologically Active Lipid Oleamide. Proc Natl Acad Sci US A. 1998;95:4102–4107. doi: 10.1073/pnas.95.8.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lees G, Dougalis A. Differential Effects of the Sleep-Inducing Lipid Oleamide and Cannabinoids on the Induction of Long-Term Potentiation in the CA1 Neurons of the Rat Hippocampus In Vitro. Brain Res. 2004;997:1–14. doi: 10.1016/j.brainres.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Yost CS, Hampson AJ, Leonoudakis D, Koblin DD, Bornheim LM, Gray AT. Oleamide Potentiates Benzodiazepine-Sensitive γ-Aminobutyric Acid Receptor Activity But Does Not Alter Minimum Alveolar Anesthetic Concentration. Anesth Analg. 1998;86:1294–1299. doi: 10.1097/00000539-199806000-00031. [DOI] [PubMed] [Google Scholar]
- 36.Huitrón-Reséndiz S, Gombart L, Cravatt BF, Henriksen SJ. Effect of Oleamide on Sleep and its Relationship to Blood Pressure, Body Temperature, and Locomotor Activity in Rats. Exp Neurol. 2001;172:235–243. doi: 10.1006/exnr.2001.7792. [DOI] [PubMed] [Google Scholar]
- 37.Boger DL, Patterson JE, Guan X, Cravatt BF, Lerner RA, Gilula NB. Chemical Requirements for Inhibition of Gap Junction Communication by the Biologically Active Lipid Oleamide. Proc Natl Acad Sci US A. 1998;95:4810–4815. doi: 10.1073/pnas.95.9.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, Gilula NB. The Sleep-Inducing Lipid Oleamide Deconvolutes Gap Junction Communication and Calcium Wave Transmission in Glial Cells. J Cell Biol. 1997;139:1785–1792. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mechoulam R, Fride E, Hanus L, Sheskin T, Bisogno T, Di Marzo V, Bayewitch M, Vogel Z. Anandamide May Mediate Sleep Induction. Nature. 1997;389:25–26. doi: 10.1038/37891. [DOI] [PubMed] [Google Scholar]
- 40.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased Seizure Susceptibility and Proconvulsant Activity of Anandamide in Mice Lacking Fatty Acid Amide Hydrolase. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to Anandamide and Enhanced Endogenous Cannabinoid Signaling in Mice Lacking Fatty Acid Amide Hydrolase. Proc Natl Acad Sci US A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional Disassociation of the Central and Peripheral Fatty Acid Amide Signaling Systems. Proc Natl Acad Sci US A. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice Lacking Fatty Acid Amide Hydrolase Exhibit a Cannabinoid Receptor-Mediated Phenotypic Hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 44.Cravatt BF, Lichtman AH. Fatty Acid Amide Hydrolase: An Emerging Therapeutic Target in the Endocannabinoid System. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 45.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaráes CRW, Jorgensen WL, Cravatt BF. Discovery of a Potent, Selective, and Efficacious Class of Reversible α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase as Analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Trifluoromethyl Ketone Inhibitors of Fatty Acid Amide Hydrolase: A Probe of Structural and Conformational Features Contributing to Inhibition. Bioorg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 47.Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Exceptionally Potent Inhibitors of Fatty Acid Amide Hydrolase: The Enzyme Responsible for Degradation of Endogenous Oleamide and Anandamide. Proc Natl Acad Sci US A. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Petrocellis L, Melck D, Ueda N, Maurelli S, Kurahashi Y, Yamamoto S, Marino G, Di Marzo V. Novel Inhibitors of Brain, Neuronal, and Basophilic Anandamide Amidohydrolase. Biochem Biophys Res Commun. 1997;231:82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- 49.Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl Arachidonyl Fluorophosphonate: A Potent Irreversible Inhibitor of Anandamide Amidase. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 50.Deutsch DG, Lin S, Hill WAG, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A. Fatty Acid Sulfonyl Fluorides Inhibit Anandamide Metabolism and Bind to the Cannabinoid Receptor. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- 51.Du W, Hardouin C, Cheng H, Hwang I, Boger DL. Heterocyclic Sulfoxide and Sulfone Inhibitors of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 2005;15:103–106. doi: 10.1016/j.bmcl.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 52.Edgemond WS, Greenberg MJ, McGinley PJ, Muthians S, Campbell WB, Hillard CJ. Synthesis and Characterization of Diazomethylarachidonyl Ketone: An Irreversible Inhibitor of N-Arachidonylethanolamine Amidohydrolase. J Pharmacol Exp Ther. 1998;286:184–190. [PubMed] [Google Scholar]
- 53.Fernando SR, Pertwee RG. Evidence that Methyl Arachidonyl Fluorophosphonate is an Irreversible Cannabinoid Receptor Antagonist. Br J Pharmacol. 1997;121:1716–1720. doi: 10.1038/sj.bjp.0701303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG. Inhibitors of Arachidonoyl Ethanolamide Hydrolysis. J Biol Chem. 1994;269 [PubMed] [Google Scholar]
- 55.Patricelli MP, Patterson JP, Boger DL, Cravatt BF. An Endogenous Sleep-Inducing Compound is a Novel Competitive Inhibitor of Fatty Acid Amide Hydrolase. Bioorg Med Chem Lett. 1998;8:613–618. doi: 10.1016/s0960-894x(98)00073-0. [DOI] [PubMed] [Google Scholar]
- 56.Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong CH, Lerner RA. Inhibition of Oleamide Hydrolase Catalyzed Hydrolysis of the Endogenous Sleep-Inducing Lipid cis-9-Octadecenamide. J Am Chem Soc. 1996;1996:5938–5945. [Google Scholar]
- 57.Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. Synthesis and Structure–Activity Relationships of FAAH Inhibitors: Cyclohexylcarbamic Acid Biphenyl Esters with Chemical Modulation at the Proximal Phenyl Ring. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 58.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, Synthesis, and Structure-Activity Relationships of Alkylcarbamic Acid Aryl Esters, a New Class of Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 59.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic Acid 3′- or 4′-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantitative Structure-Activity Relationships, and Molecular Modeling Studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 60.Muccioli GG, Fazio N, Scriba GKE, Poppitz W, Cannata F, Poupaert JH, Wouters J, Lambert DM. Substituted 2-Thioxoimidazolidin-4-ones and Imidazolidine-2,4-diones as Fatty Acid Amide Hydrolase Inhibitors Templates. J Med Chem. 2006;49:417–425. doi: 10.1021/jm050977k. [DOI] [PubMed] [Google Scholar]
- 61.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Discovery of an Exceptionally Potent and Selective Class of Fatty Acid Amide Hydrolase Inhibitors Enlisting Proteome-Wide Selectivity Screening: Concurrent Optimization of Enzyme Inhibitor Potency and Selectivity. Bioorg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 62.Hohmann AG, Suplita RL, Bolton NM, Neeley MH, Fegley D, Mangieri R, Krey J, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An Endocannabinoid Mechanism for Stress-Induced Analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 63.Alexander JP, Cravatt BF. Mechanism of Carbamate Inactivation of FAAH: Implications for the Design of Covalent Inhibitors and In Vivo Functional Probes for Enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible Inhibitors of Fatty Acid Amide Hydrolase that Promote Analgesia: Evidence for an Unprecedented Combination of Potency and Selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401.See also: Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Fatty Acid Amide Hydrolase Inhibitors Display Broad Selectivity and Inhibit Multiple Carboxylesterases as Off-Targets. Neuropharmacol. 2007 doi: 10.1016/j.neuropharm.2006.11.009. in press.
- 65.Alexander JP, Cravatt BF. The Putative Endocannabinoid Transport Blocker LY2183240 Is a Potent Inhibitor of FAAH and Several Other Brain Serine Hydrolases. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- 66.Boger DL, Miyauchi H, Hedrick MP. α-Keto Heterocycle Inhibitors of Fatty Acid Amide Hydrolase: Carbonyl Group Modification and α-Substitution. Bioorg Med Chem Lett. 2001;11:1517–1520. doi: 10.1016/s0960-894x(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 67.Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of Fatty Acid Amide Hydrolase Produces Analgesia by Multiple Mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romero FA, Hwang I, Boger DL. Delineation of a Fundamental α-Ketoheterocycle Substituent Effect For Use in the Design of Enzyme Inhibitors. J Am Chem Soc. 2006;68:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romero FA, Du W, Hwang I, Rayl TJ, Leung D, Hoover HS, Cravatt BF, Boger DL. Potent and Selective α-Ketoheterocycle-Based Inhibitors of the Anandamide and Oleamide Catabolizing Enzyme, Fatty Acid Amide Hydrolase. J Med Chem. 2007 doi: 10.1021/jm0611509. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering Potent and Selective Reversible Inhibitors of Enzymes in Complex Proteomes. Nature Biotech. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 71.Sonogashira K, Tohda Y, Hagihara N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 72.Saikachi H, Kitagawa T, Sasaki H, Van Leusen AM. Synthesis of Furan Derivatives. LXXXV. Condensation of Heteroaromatic Aldehydes with Tosylmethyl Isocyanide. Chem Pharm Bull. 1969;27:793–796. [Google Scholar]
- 73.Harn NK, Gramer CJ, Anderson BA. Acylation of Oxazoles by the Copper-Mediated Reaction of Oxazol-2-ylzinc chloride Derivatives. Tetrahedron Lett. 1995;36:9453–9456. [Google Scholar]
- 74.Dondoni A, Fantin G, Fogagnolo M, Medici A, Pedrini P. Synthesis of (Trimethylsily1)thiazoles and Reactions with Carbonyl Compounds. Selectivity Aspects and Synthetic Utility. J Org Chem. 1988;53:1748–1761. [Google Scholar]
- 75.Dondoni A, Mastellari AR, Medici A, Negrini E, Pedrini P. Synthesis of Stannylthiazoles and Mixed Stannylsilylthiazoles and Their Use for a Convenient Preparation of Mono- and Bis-Halothiazoles. Synthesis. 1986:757–760. [Google Scholar]
- 76.Pocar D, Stradi R, Trimarco P. Enamines from Cyclopropylketones. Tetrahedron. 1975;31:2427–2429. [Google Scholar]
- 77.Yovell J, Hirsch D, Sarel S. Acid–Catalyzed Addition of Secondary Amines to Cyclopropyl Ketones. Mass Spectra of Some Cyclic Aminobutyrophenones. J Org Chem. 1977;42:850–855. [Google Scholar]
- 78.Davis FA, Vishwakarma LC, Billmers JM, Finn J. Synthesis of α-Hydroxy Carbonyl Compounds (Acyloins): Direct Oxidation of Enolates Using 2-Sulfonyloxaziridines. J Org Chem. 1984;49:3241–3243. [Google Scholar]
- 79.Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF. Comparative Characterization of a Wild Type and Transmembrane Domain-Deleted Fatty Acid Amide Hydrolase: Identification of the Transmembrane Domain as a Site for Oligomerization. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
- 80.For 2b: Mwt = 334, 4 H-bond acceptors, cLogP = 4.12; For 5hh: Mwt = 369, 4 H-bond acceptors, cLogP = 4.67; for 11j: Mwt = 354, 4 H-bond acceptors, cLogP = 4.12, 5 rotatable bonds.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and characterization, FAAH assay measurement errors, and purities (HPLC analysis) of the FAAH inhibitors disclosed herein. This information is available free of charge via the Internet at http://pubs.acs.org.