

Acidic nondenaturing PAGE revealed inhomogeneity of the MASP-1 catalytic region caused by deamidation as the reason behind previous unsuccessful crystallization attempts. Monitoring the separation of the various species by acidic nondenaturing PAGE during purification helped to obtain pure protein, which was subsequently crystallized.
Keywords: innate immunity, complement-control protein domain, modular serine proteases, mannan-binding lectins, collectins, acidic PAGE
Abstract
MASP-1, a multidomain serine protease, is a component of the lectin pathway of complement. Its precise function is unknown, although it seems to enhance the complement-activating capacity of MASP-2, a related enzyme. MASP-1 has also been implicated as playing a role in blood coagulation. It is mostly found associated with mannose-binding lectin (MBL) and ficolins. Early attempts to crystallize MASP-1 failed because of the inhomogeneity of the purified material. MASP-1 was shown by acidic nondenaturing PAGE to be composed of differently charged species, which are most likely to be the products of deamidation occurring during the refolding procedure. Sequential cation-exchange and anion-exchange chromatography resulted in a homogeneous material, which was successfully crystallized. The best crystal diffracted to 2.55 Å resolution and belonged to space group P212121, with unit-cell parameters a = 68.4, b = 70.4, c = 121.4 Å. The crystal structure of MASP-1 may help in understanding the function of this mysterious serine protease.
1. Introduction
Complement, a central element of the immune system, is capable of recognizing and eliminating invading microorganisms, altered host cells and immune aggregates through opsonization or cell lysis (Law & Reid, 1995 ▶). It is a sophisticated cascade system, in which the serine protease components activate each other in a strictly ordered manner. The complement system can be activated by three routes: the classical, the lectin and the alternative pathways. The initiator molecule of the classical pathway is the C1 complex, which is composed of a C1q molecule and the C1s–C1r–C1r–C1s tetramer. C1r and C1s are serine proteases and C1q is the recognition element. Upon binding to immune complexes autoactivation of C1r occurs, which then activates C1s which cleaves C4 and C2, the upcoming components of the cascade.
The lectin pathway of complement is dependent on ficolins and mannose-binding lectin (MBL, also called mannan-binding lectin or mannan-binding protein), which bind to the carbohydrate structures of pathogens (Thiel, 2007 ▶). Initially, MBL was thought to activate complement via the action of the C1r2C1s2 tetramer (Lu et al., 1990 ▶). Subsequently, it turned out that MBL in plasma is associated with a different enzyme, termed MBL-associated serine protease or MASP (Matsushita & Fujita, 1992 ▶). Characterization of this enzyme revealed that it is actually a mixture of two proteases: the major component MASP-1 and another termed MASP-2 (Thiel et al., 1997 ▶). In contrast to the C1 complex, however, MBL forms distinct complexes with MASP-1 and MASP-2 (Chen & Wallis, 2001 ▶; Vorup-Jensen et al., 2000 ▶). There are two other MBL-associated proteins MASP-3 and MAp19, the functions of which are unknown (Sørensen et al., 2005 ▶).
MASP-2 was found to cleave both C4 and C2 efficiently, while MASP-1 only has significant activity towards C2 (Ambrus et al., 2003 ▶). The major complement-activating protease of the lectin pathway is therefore MASP-2, while MASP-1 can enhance the effect of MASP-2 (Møller-Kristensen et al., 2007 ▶). On the other hand, MASP-1 was found to cleave other substrates such as fibrinogen and factor XIII (Hajela et al., 2002 ▶) and was active on gelatin zymography (Gál et al., 2007 ▶). The physiological importance of these reactions is unclear; nevertheless, MASP-1 seems to have a broader selectivity for protein substrates than C1r, C1s and even MASP-2.
MASPs, C1r and C1s share a common domain organization: the serine protease (SP) domain is preceded by five regulatory domains (Fig. 1 ▶). The catalytic region of MASP-1 was crystallized, encompassing the first and second complement-control protein (CCP1 and CCP2) domains and the SP domain.
Figure 1.

Schematic representation of the domain structure of MASP-1 and the expressed fragment. The related proteins C1r, C1s, MASP-2 and MASP-3 share the same domain organization as MASP-1 (Gál et al., 2007 ▶).
2. Materials and methods
2.1. MASP-1 expression and refolding
The catalytic region of MASP-1 was expressed as described previously (Ambrus et al., 2003 ▶) in the form of inclusion bodies. The expressed construct was composed of an Ala-Ser-Met-Thr expression-enhancing tag followed by the Gly298–Asn699 sequence of MASP-1 comprising of the CCP1, CCP2 and SP domains. The refolding procedure was modified, which resulted in a more than tenfold increase in the yield compared with the original procedure (Ambrus et al., 2003 ▶). Inclusion bodies were dissolved in a small volume of 7 M guanidine–HCl, 50 mM Tris, 50 mM DTT pH 8.0. The concentration of MASP-1 CCP1-CCP2-SP was estimated by SDS–PAGE analysis, comparing the intensity of the MASP-1 band to purified controls. The solubilized MASP-1 was then diluted to 8 mg ml−1 with 7 M guanidine–HCl, 50 mM Tris, 50 mM DTT pH 8.0. The solubilized protein (15 ml of the 8 mg ml−1 solution) was added to 500 ml of 0.3 M arginine, 1 M NaCl, 50 mM Tris, 5 mM EDTA, 4 mM glutathione, 4 mM oxidized glutathione pH 9.4 while being stirred at room temperature. The refolding mixture (pH 9.4) was then kept at 277 K for a month. The refolding was completed by dialyzing the mixture for 2 d against 2 × 10 l of 1 mM Tris, 0.5 mM EDTA pH 8.0 with one exchange after day 1. MASP-1 autoactivates during the dialysis step, resulting in the two-chain active form. Finally, benzamidine–HCl was added to 1 mM final concentration.
2.2. Purification
In order to avoid autolysis of MASP-1 CCP1-CCP2-SP, 1 mM benzamidine–HCl was used in all buffers throughout the purification. The pH of the dialyzed refolded protein was adjusted to 6.8 with the addition of 1 M NaH2PO4. The sample was applied onto a 20 ml SP Sepharose High Performance column (GE Healthcare) and eluted with a 15-column-volume 0–300 mM NaCl gradient in 10 mM sodium phosphate, 1 mM benzamidine–HCl pH 6.8 buffer. Selected fractions were dialyzed against 5 mM Tris, 0.5 mM EDTA, 1 mM benzamidine–HCl pH 8.8 and applied onto a 20 ml Source 30Q column (GE Healthcare) equilibrated with the same buffer and eluted with a 20–100 mM NaCl gradient in 5 mM Tris, 0.5 mM EDTA, 1 mM benzamidine–HCl pH 8.8 buffer in 15 column volumes. Selected fractions were combined, supplemented with benzamidine–HCl to 20 mM final concentration and concentrated to 13 mg ml−1 using spin concentrators.
2.3. Acidic nondenaturing PAGE
The 5× sample buffer contained 50%(v/v) glycerol, 0.02%(w/v) methyl green, 0.375 M MES pH 5.3. The running-buffer composition was 200 mM β-alanine, 30 mM MES pH 5.3. The resolving gel formulation was 12.5% T, 2.67% C acrylamide/bis-acrylamide, 0.375 M MES pH 5.3, while the stacking gel contained 5% T, 2.67% C acrylamide/bis-acrylamide, 0.188 M MES pH 5.3. Samples were mixed with 1/4 volume of sample buffer and were run at 180 V for 90 min (Bio-Rad Mini-Gel format). The discontinuous buffer system was modified from Reisfeld et al. (1962 ▶).
2.4. Crystallization and X-ray diffraction analysis
MASP-1 CCP1-CCP2-SP was crystallized using the hanging-drop vapour-diffusion method at 293 K. 2 µl protein solution at 13 mg ml−1 in 50 mM NaCl, 5 mM Tris, 0.5 mM EDTA, 20 mM benzamidine–HCl pH 8.8 buffer was mixed with 2 µl reservoir solution, which contained 11%(w/v) PEG 3350, 0.1 M MES pH 6.5 and 0, 5 or 10%(v/v) glycerol. Crystals were briefly soaked in 11%(w/v) PEG 3350, 0.1 M MES pH 6.5, 20%(v/v) glycerol, mounted on nylon loops from Hampton Research and flash-cooled in liquid nitrogen. Data were collected at 100 K on the Hamburg DESY synchrotron beamline X11 at a wavelength of 0.8148 Å using a MAR 345 image-plate detector. The images were integrated and scaled using XDS and XSCALE (Kabsch, 1993 ▶).
3. Results and discussion
We first attempted to crystallize MASP-1 CCP1-CCP2-SP (Fig. 1 ▶) expressed and purified by the method published previously (Ambrus et al., 2003 ▶). However, the amount obtained was quite low and the preparation was contaminated with a breakdown product of MASP-1 (Ambrus et al., 2003 ▶). Optimization of the refolding procedure resulted in a more than tenfold increase in the yield, which provided a sufficient amount of active enzyme. Roughly 10–15 mg active MASP-1 CCP1-CCP2-SP can be produced from 120 mg protein in inclusion bodies, which is high for a three-domain protease containing eight disulfide bonds. However, we later found that purification by anion-exchange chromatography produced at least two peaks corresponding to monomeric MASP-1 CCP1-CCP2-SP. All of these forms had nearly identical enzymatic activity (data not shown) and formed covalent complexes with C1 inhibitor analogously as described in Beinrohr et al. (2007 ▶). A molecular weight in the range 45 464–45 471 Da (calculated: 45 462 Da) was found by electrospray ionization mass-spectrometric analysis for all forms. The difference is within the range of the error of the measurement. Deamidation of proteins is a common modification of proteins at high pH. Our data suggest that the relatively high pH applied (pH 9.4), which is beneficial for refolding, could have caused deamidation of MASP-1 CCP1-CCP2-SP, causing charge heterogeneity of the product. Deamidation converts a CONH2 group to a COOH, causing a change in the molecalar weight of only 1 Da. The sequence Asn-Gly, which is found twice in the expressed protein, is especially prone to deamidation (Robinson, 2002 ▶).
A sequential ion-exchange purification strategy was developed to produce homogenous material for crystallization. Avoiding auto-degradation of MASP-1 CCP1-CCP2-SP was also crucial. Purification in the presence of 1 mM benzamidine–HCl by cation-exchange chromatography produced two major peaks on top of a broader peak caused by benzamidine (Fig. 2 ▶ a). Owing to its basic nature (pI 7.6), analysis by conventional native PAGE (using the Laemmli buffer system without SDS) was not feasible. Analysis by nondenaturing acidic PAGE revealed three slightly differently migrating species (Fig. 2 ▶ b). Deamidation adds a negative charge to the protein, making it migrate more slowly on acidic gels. The fastest migrating protein is presumably unmodified MASP-1 CCP1-CCP2-SP. From a cation-exchange column the unmodified protein is eluted last, while from an anion-exchange column it is eluted first. By carefully selecting and analyzing fractions, pure homogeneous MASP-1 CCP1-CCP2-SP was obtained that could be crystallized successfully.
Figure 2.

(a) Separation by cation-exchange chromatography. Peaks I and II contain MASP-1 CCP1-CCP2-SP, while the broad peak labelled ‘Bz’ is caused by benzamidine–HCl. The absorbance of benzamidine partially masks the protein peaks. The solid line is the absorbance at 280 nm, the dashed line is the absorbance at 254 nm and the dotted line is the conductivity. (b) Analysis of chromatographic fractions by native acidic PAGE. Peak I (lane 1) and peak II (lane 2) of the cation-exchange step are a mixture of two species each and three overall. Peak II was further purified by anion-exchange chromatography. The purest fractions contained a single species (lane 3) of MASP-1 CCP1-CCP2-SP.
The largest crystals grew from solutions with no glycerol present to dimensions of up to 0.2 × 0.2 × 2 mm. However, these large crystals did not withstand soaking in the cryoprotectant solution, which was 11%(w/v) PEG 3350, 0.1 M MES pH 6.5 and 20%(v/v) glycerol. Soaking in different cryosolutions with MDP or PEG 400 as cryoprotectant and soaking in Paratone oil (Hampton Research) were similarly unsuccessful. Smaller crystals, especially those which grew in the presence of 10%(v/v) glycerol, could tolerate brief (2–5 s) soaking into the cryosolution containing 20%(v/v) glycerol. Gradual soaking by increments of 5%(v/v) glycerol was also beneficial. The ends of these smaller crystals appeared to have minor cracks under the microscope in cryoprotectant solution; however, some diffracted well. The better diffracting crystals had typical dimensions of about 0.07 × 0.07 × 0.7 mm (Fig. 3 ▶). Finally, a complete 2.55 Å resolution data set was collected from a single crystal. The crystals belong to the orthorhombic P212121 space group and have unit-cell parameters a = 68.4, b = 70.4, c = 121.4 Å, one molecule per asymmetric unit and approximately 62% solvent content (Matthews, 1968 ▶). Data-collection and processing statistics are presented in Table 1 ▶. Molecular replacement using the structures of individual CCP domains of MASP-2 (Gál et al., 2005 ▶) and SP domains of homologous serine protease structures gave reasonable solutions. Currently, structure determination and model building are in progress.
Figure 3.

Typical crystals of MASP-1 CCP1-CCP2-SP. Crystals appeared after 2–3 d in drops equilibrated against 11%(w/v) PEG 3350, 0.1 M MES pH 6.5 and were harvested after 5 d.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 68.4, b = 70.4, c = 121.4 |
| Crystal-to-detector distance (mm) | 430 |
| Maximum resolution (Å) | 2.55 |
| Rotation per exposure (°) | 0.3 |
| Time per image (s) | 40 |
| Total rotation for data set (°) | 150 |
| Resolution range (Å) | 5.98–2.55 (2.616–2.55) |
| No. of observations | 114297 (6216) |
| No. of unique reflections | 19349 (1066) |
| Matthews coefficient (Å3 Da−1) | 3.22 |
| Rmeas† | 10.4 (51.5) |
| Completeness (%) | 97.9 (97.4) |
| Average I/σ(I) | 15.96 (4.35) |
| Wilson B (Å2) | 30.892 |
R
meas = 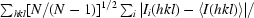
 .
.
Crystal structures of related active fragments of C1r, C1s and MASP-2 have been determined (Kardos et al., 2008 ▶; Gaboriaud et al., 2000 ▶; Harmat et al., 2004 ▶) and also that of their principal inhibitor C1 inhibitor (Beinrohr et al., 2007 ▶). Analysis of the protein and DNA sequence and the genomic organization of MASP-1 revealed that its SP domain differs from those of other members of the family with the same domain organization (Fig. 1 ▶). The MASP-1 SP domain is encoded by six exons (opposed to one), the active serine is encoded by a TCN codon (as opposed to AGY) and the histidine loop found in trypsin/chymotrypsin is also present in MASP-1 (Sørensen et al., 2005 ▶). Structural alignment of the SP domain of active MASP-1 with those of C1r, C1s, MASP-2, thrombin and trypsin will help us to reveal the structural basis of the broader substrate specificity of MASP-1 compared with those of the other members of the C1r/C1s/MASP family. Although the exact functions of MASP-1 remain to be identified, the structure will take us a step closer to understanding the role of this intriguing enzyme.
Acknowledgments
We thank Dr Katalin Kékesi (ELTE, Budapest, Hungary) for the MS analysis of MASP-1 species. We thank Dr Michele Cianci (EMBL, Hamburg, Germany) for help during data collection. Access to EMBL beamline X11 at the DORIS storage ring, DESY, Hamburg is gratefully acknowledged. Data collection was supported by a travel grant from the European Community (Research Infrastructure Action under the FP6 ‘Structuring the European Research Area Specific Programme’ to the EMBL Hamburg Outstation, Contract No. RII3-CT-2004-506008). This work was supported by the Ányos Jedlik grant NKFP_07_1-MASPOK07 from the Hungarian National Office for Research and Technology (NKTH) and by Hungarian Scientific Research Fund (OTKA) grants NI61915, F67937 and NK67800.
References
- Ambrus, G., Gál, P., Kojima, M., Szilágyi, K., Balczer, J., Antal, J., Gráf, L., Laich, A., Moffatt, B. E., Schwaeble, W., Sim, R. B. & Zavodszky, P. (2003). J. Immunol.170, 1374–1382. [DOI] [PubMed]
- Beinrohr, L., Harmat, V., Dobó, J., Lörincz, Z., Gál, P. & Závodszky, P. (2007). J. Biol. Chem.282, 21100–21109. [DOI] [PubMed]
- Chen, C. & Wallis, R. (2001). J. Biol. Chem.276, 25894–25902. [DOI] [PubMed]
- Gaboriaud, C., Rossi, V., Bally, I., Arlaud, G. J. & Fontecilla-Camps, J. C. (2000). EMBO J.19, 1755–1765. [DOI] [PMC free article] [PubMed]
- Gál, P., Barna, L., Kocsis, A. & Závodszky, P. (2007). Immunobiology, 212, 267–277. [DOI] [PubMed]
- Gál, P., Harmat, V., Kocsis, A., Bián, T., Barna, L., Ambrus, G., Végh, B., Balczer, J., Sim, R. B., Náray-Szabó, G. & Závodszky, P. (2005). J. Biol. Chem.280, 33435–33444. [DOI] [PubMed]
- Hajela, K., Kojima, M., Ambrus, G., Wong, K. H., Moffatt, B. E., Ferluga, J., Hajela, S., Gál, P. & Sim, R. B. (2002). Immunobiology, 205, 467–475. [DOI] [PubMed]
- Harmat, V., Gál, P., Kardos, J., Szilágyi, K., Ambrus, G., Végh, B., Náray-Szabó, G. & Závodszky, P. (2004). J. Mol. Biol.342, 1533–1546. [DOI] [PubMed]
- Law, S. K. A. & Reid, K. B. M. (1995). Complement, 2nd ed. Oxford: IRL Press.
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kardos, J., Harmat, V., Palló, A., Barabás, O., Szilágyi, K., Gráf, L., Náray-Szabó, G., Goto, Y., Závodszky, P. & Gál, P. (2008). Mol. Immunol.45, 1752–1760. [DOI] [PubMed]
- Lu, J., Thiel, S., Wiedemann, H., Timpl, R. & Reid, K. B. M. (1990). J. Immunol.144, 2287–2294. [PubMed]
- Matsushita, M. & Fujita, T. (1992). J. Exp. Med.176, 1497–1502. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Møller-Kristensen, M., Thiel, S., Sjöholm, A., Matsushita, M. & Jensenius, J. C. (2007). Int. Immunol.19, 141–149. [DOI] [PubMed]
- Reisfeld, R. A., Lewis, U. J. & Williams, D. E. (1962). Nature (London), 195, 281–283. [DOI] [PubMed]
- Robinson, N. E. (2002). Proc. Natl Acad. Sci. USA, 99, 5283–5288.
- Sørensen, R., Thiel, S. & Jensenius, J. C. (2005). Springer Semin. Immunopathol.27, 299–319. [DOI] [PubMed]
- Thiel, S. (2007). Mol. Immunol.44, 3875–3888. [DOI] [PubMed]
- Thiel, S., Vorup-Jensen, T., Stover, C. M., Schwaeble, W., Laursen, S. B., Poulsen, K., Willis, A. C., Eggleton, P., Hansen, S., Holmskov, U., Reid, K. B. & Jensenius, J. C. (1997). Nature (London), 386, 506–510. [DOI] [PubMed]
- Vorup-Jensen, T., Petersen, S. V., Hansen, A. G., Poulsen, K., Schwaeble, W., Sim, R. B., Reid, K. B. M., Davis, S. J., Thiel, S. & Jensenius, J. C. (2000). J. Immunol.165, 2093–2100. [DOI] [PubMed]