

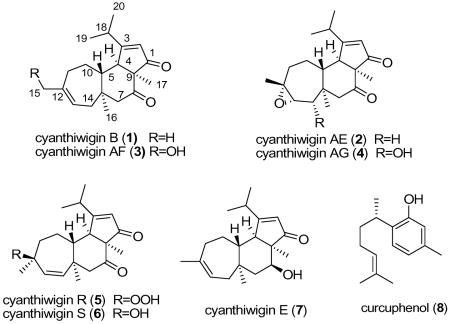
Abstract
Microbial transformation studies of the marine diterpene cyanthiwigin B (1), isolated from the Jamaican sponge Myrmekioderma styx, were accomplished. Two actinomycetes cultures Streptomyces NRRL 5690 and Streptomyces spheroides, significantly metabolized cyanthiwigin B to new metabolites. Streptomyces NRRL 5690 transformed cyanthiwigin B to three new compounds cyanthiwigins AE (2), AF (3), AG (4), and the known cyanthiwigin R (5). S. spheroides transformed cyanthiwigin B to cyanthiwigins S (6), E (7), and AE (2). All microbial metabolized derivatives (2 ~ 7) of cyanthiwigin B exhibited the ability to increase the antimicrobial activity of curcuphenol, the major antimicrobial sesquiterpene isolated from M. styx.
Cyanthiwigins are a class of 5,6,7-tricarbocyclic diterpenes, which were first isolated from a Jamaican sponge Epipolasis reiswigi in 1992.1 Cyanthiwigin C and two analogous epoxides were reported in the same year from a Venezuelan sponge Myrmekioderma styx.2 Since this diterpene class has the same tricarbocyclic skeleton as the cyanthins, metabolites from the bird’s nest fungus Cyanthus sp.,3 they were named cyanthiwigins. In the interest of identifying anticancer and anti-infective leads from Jamaican deep reef invertebrates, we isolated thirty diterpenes, cyanthiwigins A-AD, from the sponge Myrmekioderma styx,4,5 of which cyanthiwigin B (1) is presented in the highest yield. Additionally, cyanthiwigin B has the unique characteristic of increasing the antimicrobial activity of curcuphenol (8), a sesquiterpene isolated from the same sponge.6 Recently, total synthesis of cyanthiwigin U was achieved.7 Microbial transformations have been shown to be a productive method for the modification of natural products.8 In order to improve the synergetic antimicrobial effect, cyanthiwigin B (1) was subjected to a biotransformation study.
Biotransformations of terpenoids have been reported as having particular significance due to their application as fragrances or flavoring agents, their antibacterial, antiviral or cytotoxic activities and their increasing importance as specific pharmacological agents. The ability of microbial transformations to introduce functional groups with regio- and stereo-selectivity remains unrivaled by existing chemical methodologies.9 In most cases biotransformation leads to the inactivation of drugs. However the biotransformation can also lead to the generation of a more active or less toxic metabolites. In some instances, biotransformations can produce reactive or toxic intermediates with potential toxicological implications. Hence, an understanding of the metabolism of new chemical entities is a valuable component of the drug discovery process.10
Results and Discussion
Ten microorganisms were screened for their ability to transform cyanthiwigin B (1) to more active metabolites. Two actinomycetes cultures Streptomyces NRRL 5690 and Streptomyces spheroides, showed significant biocatalysis of cyanthiwigin B and were selected for preparative-scale fermentation. Streptomyces NRRL 5690 was able to transform cyanthiwigin B to cyanthiwigins AE (2, 13.5%), AF (3, 3%), AG (4, 3%), and R (5, 2%). S. spheroides transformed cyanthiwigin to cyanthiwigins S (6, 2%), E (7, 1%) and AE (2, 1.5%).
Cyanthiwigin AE (2) was obtained as a white powder. Its molecular formula was deduced as C20H28O3 by high-resolution ESIMS [M+Na] 339.1918 (calcd. 339.1936). The 1H NMR provided characteristic signals of cyanthiwigin type diterpenes, including an olefinic proton at δ 5.81 (H-2), a methine proton at δ 2.73 (d, J=8.9 Hz, H-4), and five methyl protons at δ 0.94 (s), 1.16 (s), 1.22 (s), 1.19 (d, J=7.0 Hz), and 1.29 (d, J=6.5 Hz). In comparison to cyanthiwigin B (1), 2 revealed the absence of carbon signals of the double bound between C-12 and C-13 and new carbon signals at δ 60.0 and 62.3, which indicated the double bound was converted to an epoxy functionality. The structure was verified by long-range heteronuclear correlations (HMBC) of δ 2.03 (H-11), 1.88 (H-14), 1.22 (H-15) to δ 60.0 (C-12) and 62.3 (C-13), as well as δ 2.84 (H-13) to δ 41.3 (C-6,14), 60.0 (C-12) and 24.5 (C-15). The 1D and 2D NMR data for the remainder of the structure were superimposable with that of cyanthiwigin B (1). The relative stereochemistry of the epoxide group was assigned as α by comparison of the coupling constant and NOE correlations of H-13 with those of cyanthiwigins H and J.4 The signal of H-13 was shown to be a triplet with a small 3J coupling constant of 3.6 Hz for cyanthiwigin AE, which is significantly different as compared to the large 3J values of 8.0 and 7.2 Hz for cyanthiwigins H and J. In the NOESY spectrum, H-13 (δ 2.84) did not show correlations to H-16 (δ 0.94), while the NOE correlations between H-13 and H-16 were observed in cyanthiwigins H and J.4 These indicated the α orientation of the epoxide group of cyanthiwigin AE rather than the β orientation for cyanthiwigins H and J. To confirm the stereochemistry, crystals of 2 were grown in acetone. X-ray diffraction analysis was performed and the crystal structure with relative configuration is shown in Fig. 1.
Figure 1.
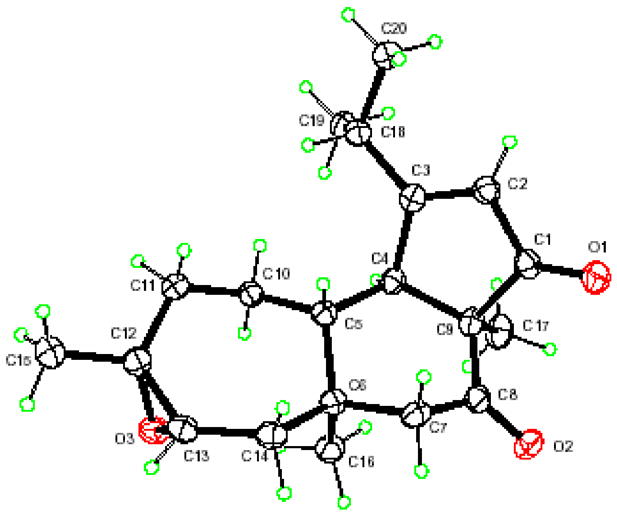
X-ray structure of 2
Cyanthiwigin AF (3) was also obtained as a white powder. The molecular formula C20H28O3 was shown by high-resolution ESIMS [M+Na] 339.1927 (calcd. 339.1936) to have one more oxygen than that of cyanthiwigin B (1). The 1H NMR spectral data of 3 were comparable with that of cyanthiwigin B with the exception of the loss of a methyl proton signal at δ 1.74 (H-15) and the addition of an oxygenated methylene proton at δ 3.94, which suggested a hydroxyl group at C-15. The 13C NMR spectrum revealed the hydroxymethylene carbon at δ 66.8, which was confirmed to be at C-15 by long-range heteronuclear correlation with H-11 (δ 2.19).
Cyanthiwigin AG (4) was isolated as a white powder and its molecular formula C20H28O4 was obtained from the high-resolution ESIMS [M+Na] 355.1854 (calcd. 355.1885), which has the same degree of unsaturation but two more oxygen atoms than cyanthiwigin B (1). The 1H and 13C NMR data (see Table 1) showed that cyanthiwigin AG is both a hydroxylated and epoxidized derivative of cyanthiwigin B. The epoxide functionality, δ H 3.06, δ C 58.8 and 69.7, was located between C-12 and C-13 based on the HMBC correlations between δ 1.24 (H-15), 2.05 (H-11α), 1.61 (H-11β) and C-12 and C-13. The hydroxyl group was assigned to C-14 by the HMBC correlations from δ 0.95 (H-16) to δ 74.4 (C-14) and δ 3.66 (H-14) to δ 46.1 (C-6) and 14.2 (C-16). The rest of the molecule remained unmetabolized. A correlation between H-14 and H-5 was observed in the NOESY experiment, indicating that H-14 has the same β orientation with H-5. The broad singlet of the H-13 signal suggested a cis axial-equatorial relationship of H-13 and H-14. H-13 has NOE correlations with H-14 and H-15, indicating the β orientation of H-13 and CH3-15 and the α orientation of the epoxy group.
Table 1.
1H and 13C NMR Data of cyanthiwigins AF and AG (2~ 4)a
| 2
|
3
|
4
|
||||
|---|---|---|---|---|---|---|
| δH( J in Hz) | δC, mult. | δH ( J in Hz) | δC, mult. | δH( J in Hz) | δ C, mult. | |
| 1 | 204.3, qC | 204.2, qC | 203.7, qC | |||
| 2 | 5.81, s | 122.4, CH | 5.84, s | 122.7, CH | 5.81, s | 122.2, CH |
| 3 | 192.9, qC | 192.9, qC | 192.6, qC | |||
| 4α | 2.73, d (8.9) | 58.9, CH | 2.71, d (9.2) | 58.7, CH | 2.76, d (8.4) | 58.9, CH |
| 5β | 1.50, t (8.8) | 52.5, CH | 1.77, t (9.6) | 56.5, CH | 1.45, t (9.2) | 49.0, CH |
| 6 | 41.3, qC | 38.8, qC | 46.1, qC | |||
| 7α | 1.91, d (13.0) | 56.1, CH2 | 1.91, d (13.6) | 56.3, CH2 | 1.85, d (12.4) | 51.5, CH2 |
| 7β | 2.10, d (13.0) | 2.15, d (13.6) | 2.67, d (12.4) | |||
| 8 | 204.0, qC | 204.7, qC | 204.2, qC | |||
| 9 | 63.1, qC | 63.4, qC | 62.8, qC | |||
| 10α | 1.81, m | 27.3, CH2 | 1.51, m | 28.7, CH2 | 1.89, m | 27.6, CH2 |
| 10β | 1.73, m | 2.03, m | 2.04, m | |||
| 11α | 2.03, m | 33.7, CH2 | 2.01, m | 38.5, CH2 | 2.05, m | 33.9, CH2 |
| 11β | 2.19, m | 1.61, dt (1.2, 15.2) | ||||
| 12 | 60.0, qC | 145.5, qC | 58.8 qC | |||
| 13 | 2.85, t (3.6) | 62.3, CH | 5.57, m | 121.2, CH | 3.06, br. s | 69.7, CH |
| 14α | 1.88, br, s | 41.3, CH2 | 1.86, dd (8.8, 14.4) | 42.0, CH2 | 3.66, d (5.6) | 74.4, CH |
| 14β | 2.22, m | |||||
| 15 | 1.22, s | 24.5, CH3 | 3.94, s | 66.8, CH2 | 1.24, s | 24.9, CH3 |
| 16 | 0.94, s | 19.6, CH3 | 0.72, s | 17.5, CH3 | 0.95 | 14.2, CH3 |
| 17 | 1.16, s | 23.9, CH3 | 1.19, s | 24., CH3 | 1.16, s | 23.6, CH3 |
| 18 | 2.87, m | 30.5, CH | 2.91, m | 31.2, CH | 2.85, m | 29.9, CH |
| 19 | 1.29, d (6.5) | 21.0, CH3 | 1.29, d (6.8) | 20.7, CH3 | 1.30, d (6.8) | 20.8, CH3 |
| 20 | 1.19, d (6.5) | 21.3, CH3 | 1.18, d (6.8) | 21.3, CH3 | 1.20, d (6.8) | 21.2, CH3 |
| OH | 3.79, br, s | 4.45, d ( 5.6) | ||||
1H NMR at 400 MHz, 13C NMR at 100MHz, referenced to residual solvent acetone-d6.
Compounds 5–7 were identified as cyanthiwigins R, S, and E by comparison of the TLC and NMR data with the authentic samples isolated from the same sample of sponge from which cyanthiwigin B was isolated.4 This data suggested that cyanthiwigins R, S, and E, originally isolated from the sponge M. styx, maybe metabolites of sponge associated microbes.
All microbial metabolites (2–7) of cyanthiwigin B were tested for their ability to increase the anti-microbial activity of curcuphenol (8) and the results are shown in Table 2. Cyanthiwigins were first tested for the antimicrobial activity, and then mixed with curcuphenol followed by the repeat testing for antimicrobial activity. Due to the limited amount of cyanthiwigins AF (3) and AG (4), they were directly mixed with curcuphenol. Curcuphenol exhibited anti-microbial activity with IC50 15μg/mL against Cryptococcus neoformans and IC50 > 15μg/mL against Candida albicans, Staphylococcus aureus, and methicillin-resistant S. aureus (MRS). Cyanthiwigins B (1), AE (2), R (5), S (6), and E (7) were not active against all the tested microbes when assayed alone. All mixtures exhibited better activity than curcuphenol, indicating the ability of cyanthiwigins to increase the antimicrobial activity. Cyanthiwigin AE (2) and AF (3) are more potent than cyanthiwigin B (1), E (7), and S (6), and cyanthiwigin R (5) has the least potency.
Table 2.
Antimicrobial Activity
| Antifungal IC50 (μg/mL) | Antibacterial IC50 (μg/mL) | |||
|---|---|---|---|---|
| Candida albicans | Cryptococcus neoformans | Staphylococcus aureus | MRSa | |
| curcuphenol | >15 | 15 | >15 | >15 |
| cyanthiwigin B | NA | NA | NA | NA |
| cyanthiwigin B+ curcuphenol 1.1:1.8 | >15 | 6.0 | 6.5 | 7.0 |
| cyanthiwigin E | NA | NA | NA | NA |
| cyanthiwigin E+ curcuphenol 0.7:1.7 | >15 | 6.0 | 6.5 | 6.5 |
| cyanthiwigin R | NA | NA | NA | NA |
| cyanthiwigin R+ curcuphenol 1.0:1.8 | >15 | 10 | >15 | >15 |
| cyanthiwigin S | NA | NA | NA | NA |
| cyanthiwigin S+ curcuphenol 0.7:1.8 | 15 | 6.5 | 8.0 | 7.5 |
| cyanthiwigin AE | NA | NA | NA | NA |
| cyanthiwigin AE+ curcuphenol 0.6:1.8 | 15 | 6.0 | 6.5 | 6.5 |
| cyanthiwigin AF+ curcuphenol 0.5:1.8 | 15 | 5.0 | 6.5 | 6.0 |
| cyanthiwigin AG+ curcuphenol 0.5:1.8 | >15 | 7.5 | 8.5 | 9.5 |
MRS= methicillin-resistant Staphylococcus aureus.
IC50 values for curcuphenol and cyanthiwigin mixtures were calculated based on the mass of curcuphenol.
Experimental Section
General Experiment Procedures
Optical rotations were measured with a JASCO DIP-370 digital polarimeter. IR and UV spectra were obtained using an AATI Mattson Genesis Series FTIR and a Hewlett Packard 8452A diode array spectrometer. NMR spectra were measured on Bruker Avance DRX-400. 1H and 13C-NMR spectra were measured and reported in ppm by using the residual solvent peak as an internal standard (acetone-d6, δ 2.05 for proton, δ 206.7 for carbon). ESI-FTMS analyses were measured on a Bruker-Magnex BioAPEX 30es ion cyclotron HR-FT spectrometry by direct injection into an electrospray interface. HPLC was carried out on a Waters 510 model system and a dual wavelength detector.
X-ray Analysis
The crystal was colorless orthorhombic, space group P212121 with cell dimensions a = 8.474 (3) Å, b = 10.392 (3) Å, c = 19.749 (8) Å, V = 1739.1 (11) Å3, Z = 1, Dx = 1.208 Mgm−3. Intensity data for cyanthiwigin AE (2)11 were collected at 102 K on a Nonius KappaCCD (with Oxford Cryostream) diffractometer equipped with Mo Kα(λ = 0.71073 Å) radiation usingω-2θ scan, 2θ range 2.5–27.5°. 2266 independent reflections were obtained with 1871 reflections having I >2σ(I). Data reduction was by DENZOSMN11 and SCALEPAK.12 The structure was solved by direct methods and refined by full-matrix least-squares using SHELXL.13 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were constrained in calculated positions. Atomic coordinates, crystal data, final R values, and other details are included in Tables 3, 4, and 5, Supporting Information.
Material
Cyanthiwigin B (1) was isolated from Myrmekioderma styx and its structure was determined by spectral analysis as previously reported.4
Microorganisms
Streptomyces NRRL 5690, S. argenteolus. S. seokies, S. spheroides, Saccahromyces cerevisiae, Penicillium sp, Aspergillus sp, Geotrichum sp, Fusarium solani and F. oxysporium. All microorganisms were obtained from the collection at the Department of Pharmacognosy, The University of Mississippi.
Microbial Metabolism
All microorganisms were subjected to screening for their ability to transform cyanthiwigin B by inoculating them in 25 mL media that contain 3 mg cyanthiwigin B. The cultures were shaken in an incubator shaker at 200 rpm, 28°. The biotransformation process was monitored at 3, 5, 10, 14 and 18 days using TLC. Streptomyces NRRL 5690 was able to metabolize the compound in 5 days while S. spheroides metabolized the compounds from 5 to 10 days. The rest of the microorganisms did not metabolize the compound until the 18 days fermentation. For scaling-up Streptomyces NRRL 5690 and S. spheroides were grown in 100 mL media containing 20 mg of cyanthiwigin B. The cultures were shaken in an incubator shaker at 200 rpm, 28°. The fermentation was stopped after 5 days for Streptomyces NRRL 5690 and 10 days for S. spheroides.
Isolation
The whole culture of Streptomyces NRRL 5690 was extracted with EtOAc. The extract (15.9 mg) was subjected to HPLC using a Phenomenex Luna C8 column, 5 μm, 250 × 21.5 mm, eluted with a gradient of 50% acetonitrile in H2O to 100% acetonitrile over 50 min at a flow rate of 6 mL/min and a detector wavelength of 254 nm. Fraction 9 and 13 gave pure cyanthiwigin AE (2, 2.7 mg) and R (5, 0.4 mg), respectively. Fraction 5 was separated by silica gel preparative thin layer chromatography using hexane-EtOAc 1:1 as a developing solvent to yield cyanthiwigin AF (3, 0.6 mg) and AG (4, 0.6 mg).
The whole extract of Streptomyces spheroides was extracted with EtOAc. The extract (13.2 mg) was chromatographed using an HPLC column using the same condition as above. Fraction 9 and 17 gave cyanthiwigins S (6, 0.4 mg) and E (7, 0.2 mg). Fraction 13 yielded cyanthiwigin AE (2, 0.3 mg) after purification by PTLC (silica gel, hexanes-EtOAc 1:1).
Cyanthiwigin AE (2)
Colorless crystal; [α]26D –285 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 236 (4.15) nm; IR (film) νmax 2971, 1725, 1689, 1605, 1433, 1383, 1261, 1171, 870 cm−1; HRESIMS m/z339.1918 [M+Na]+ (calcd for C20H28O3Na 339.1936), 655.3964 [2M+Na]+ (calcd for C40H56O6Na 655.3975).
Cyanthiwigin AF (3)
white powder; [α]26D -284 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 236 (4.00) nm; IR (film) νmax 3448, 2963, 1724, 1687, 1603, 1455, 1384, 1260, 1086, 868 cm−1; HRESIMS m/z 339.1927 [M+Na]+ (calcd for C20H28O3Na 339.1936), 655.3981 [2M+Na]+ (calcd for C40H56O6Na 655.3975).
Cyanthiwigin AG (4)
white powder; [α]26D –145 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 236 (3.86) nm; IR (film) νmax 3436, 2964, 1725, 1691, 1605, 1454, 1259, 1006, 826 cm−1; HRESIMS m/z 355.1854 [M+Na]+ (calcd for C20H28O4Na 355.1885), 687.3824 [2M+Na]+ (calcd for C40H56O8Na 687.3872).
Supplementary Material
X-ray analysis and crystal data for cyanthiwigin AE (2). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge S. C. Sanders and B. G. Smiley from the National Center for Natural Products Research for antimicrobial testing. This work was supported by NIH grant (1R01A136596) and PharmaMar.
References and Notes
- 1.Green D, Goldberg I, Stein Z, Ilan M, Kashman Y. Nat Prod Lett. 1992;1:193–199. [Google Scholar]
- 2.Sennett SH, Pomponi SA, Wright AE. J Nat Prod. 1992;55:1421–1429. doi: 10.1021/np50088a006. [DOI] [PubMed] [Google Scholar]
- 3.Ayer WA, Taube H. Can J Chem. 1973;51:3842–3853. [Google Scholar]
- 4.Peng J, Walsh K, Weedman V, Braude IA, Kelly M, Hamann MT. Tetrahedron. 2002;58:7809–7819. [Google Scholar]
- 5.Peng J, Avery MA, Hamann MT. Org Lett. 2003;5:4575–4578. doi: 10.1021/ol035592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng J, Franzblau SG, Zhang F, Hamann MT. Tetrahedron Lett. 2002;43:9699–9702. [Google Scholar]
- 7.Pfeiffer MWB, Phillips AJ. J Am Chem Soc. 2005;127:5334–5335. doi: 10.1021/ja0509836. [DOI] [PubMed] [Google Scholar]
- 8.El Sayed KA, Yousaf M, Hamann MT, Avery MA, Kelly M, Wipf P. J Nat Prod. 2002;65:1547–1553. doi: 10.1021/np020213x. [DOI] [PubMed] [Google Scholar]
- 9.Azerad R. In: Stereoselective Biocatalysis. Patel RN, editor. Marcel Dekker, Inc; New York: 2000. pp. 153–180. [Google Scholar]
- 10.Kumar GN, Surapaneni S. Med Res Review. 2001;21:397–411. doi: 10.1002/med.1016. [DOI] [PubMed] [Google Scholar]
- 11.A full list of crystallographic data and parameters is deposited at the Cambridge Crystallographic Data Centre,12 Union Rd., Cambridge CB2 1EZ, Cambridge, UK
- 12.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 13.Sheldrick GM. SHELXL97. Program for the Refinement of Crystal Structures. University of Gottingen; Germany: 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
X-ray analysis and crystal data for cyanthiwigin AE (2). This material is available free of charge via the Internet at http://pubs.acs.org.