

Abstract
To investigate nucleic acids with selenium derivatization for crystallography, we report the first synthesis of 2′-methylseleno-thymidine phosphoramidite and its incorporation into DNAs and RNAs by solid-phase synthesis with over 99% coupling yield. The d(GTSeGTACAC)2 crystal structure was also determined at 1.40 Å resolution using Se phasing, revealing that this Se derivatization did not cause significant structure perturbation, consistent with our UV-melting study. In addition, we observed that the Se modification largely facilitated the crystallization.
Nucleic acids play key roles in genetic information storage, expression and translation, and have attracted intense research attention for a long time1-4. X-ray crystallography has been widely used to study structure and function of nucleic acid molecules.5-6 To facilitate crystal structure determination of nucleic acids, especially using Multiwavelength Anomalous Dispersion (MAD) or Single-Wavelength Anomalous Dispersion (SAD) phasing,7 Huang, Egli and coworkers were the first to develop the selenium derivatization strategy.8-13 So far, the 2′-Se derivatization has been introduced to U and C by us, and to A and G by other groups.9-15 Using the Se derivatization and phasing, research laboratories have already reported several crystal structures of DNA and RNA molecules,10,14-16including the Homo-DNA structure, otherwise the phasing of which proved extremely challenging.16
Natural tRNAs and rRNAs are heavily modified, and over 100 different modifications have been observed.17 Ribothymidine (rT) is a common modification in mature tRNAs and rRNAs. Though the ribothymidine is the invariable modification at position 54 of tRNAs of nearly all bacteria and enkarya,17b its function in the TΨC loop has not been well understood. Since our previous study indicated that the Se derivatization at the 2′-positions of the nucleotides does not cause significant perturbation of RNAs,9-11 the Se incorporation at the 2′-position of ribothymidine in tRNAs and rRNAs will help to obtain insight into the important rT modification. Furthermore, the Se incorporation at the same 2′-position of thymidine will help to study structure and function of DNA molecules. Therefore, there is an urgent need to synthesize 2′-Se-thymidine derivative, as an analog of both ribothymidine and thymidine for the RNA and DNA studies. We report here the first synthesis of the 2′-methylseleno-thymidine derivative and the corresponding phosphoramidite, its incorporation into DNAs and RNAs by solid-phase synthesis, and the UV melting and X-ray crystal structure studies of the Se-derivatized DNAs and RNAs.
The synthesis of the 2′-methylseleno-ribothymidine derivative (4) started from the glycosylation of the protected D-ribose (1) with thymine under the Vorburggen conditions.18 Intermediate 3 was synthesized by minor modifications of the literature procedures.19 In brief, due to the 2′-neighboring group effect, this glycosylation generated exclusively the protected β-ribothymidine derivative, followed by the NaOMe treatment to give fully-deprotected ribothymidine (2) in 88% yield over two steps. After the intramolecular cyclization of 2 in DMF, the 2,2′-anhydrothymidine was selectively protected with DMTr group at the 5′-position to give 3 in 87% yield in two steps. In the Se functionality incorporation step, we initially encountered problems of low yield and long reaction time in the Se introduction into anhydroribothymidine 3. We attempted to increase the reaction yield by protecting the 3′-hydroxyl group and using different reducing reagents, such as LiAlH4 and DIBAH, to form the methyl selenide nucleophile via the reduction of dimethyl diselenide. Unfortunately, we observed the nucleobase reduction and the elimination reaction. These problems were later overcome by using a weaker reducing reagent (NaBH4) at a higher temperature (50 °C) in EtOH, which gave 82% yield in three hours. Finally, the 2′-Se-ribothymidine (4) was converted to the corresponding phosphoramidite (5) in 90% yield.11a,c
Similar to solid-phase oligonucleotide synthesis using the thymidine phosphoramidite,9,11a,c this novel 2′-Se-ribothymidine phosphoramidite was incorporated into several DNA and RNA oligonucleotides under the standard conditions in the presence of 5-BMP as the coupling reagent. Analogous to our previous report, the 2′-Se functionality on rT was stable under the mild I2 treatment (20 mM, 20 seconds).9,11c The synthesized oligonucleotides were cleaved off the solid support and fully deprotected with concentrated ammonia at 55 °C overnight, followed by reversed-phase HPLC purification twice: DMTr-on and DMTr-off purification. A typical HPLC profile of the Se-DNAs with the DMTr groups is shown in Figure 1A, which is almost identical to that of the corresponding native DNA (data not shown). The HPLC analysis of the native and modified DNA octamers indicated that the Se-modified and non-modified phosphoramidites had the same incorporation efficiency, revealing a high coupling yield for the Se-T phosphoramidite (99%) as the total formation of the short abortive oligonucleotides was less than 5%. A representative HPLC purification profile of the DMTr-off Se-DNAs is shown in Figure 1B. All of the purified Se-oligonucleotides were also confirmed by MALDI-TOF MS analysis. A typical MS spectrum of the Se-oligonucleotides is shown in Figure 2; all MS data of Se-DNAs and Se-RNAs are collected in Table 1.
Figure 1.
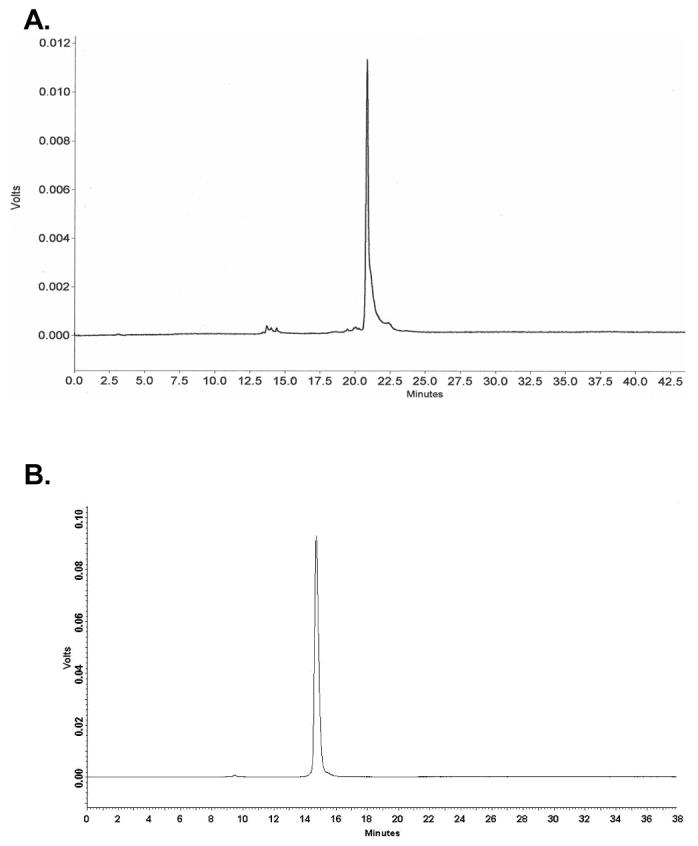
Reversed-phase HPLC analysis and purification of the Se-DNAs. A) the HPLC analysis profile of the crude DMTr-on Se-DNA (5′-DMTr-GTSeGTACAC-3′) after cleavage from the solid support and deprotection. The HPLC analysis was performed on a Zorbax SB-C18 column (4.6 × 250 mm) with a linear gradient from buffer A (20 mM triethylammonium acetate, pH 7.1) to 70% buffer B (50% acetonitrile, 20 mM triethyl ammonium acetate, pH 7.1) in 20 minutes; the Se-DNA retention time was 20.6 min. B) the HPLC purification profile of the DMTr-off Se-DNA (5′-GTSeGTACAC-3′). The sample was eluted on a Zorbax SB-C18 column (21 × 250 mm; flow rate: 6 mL/min) with the same gradient and buffers; its retention time is 14.8 min.
Figure 2.
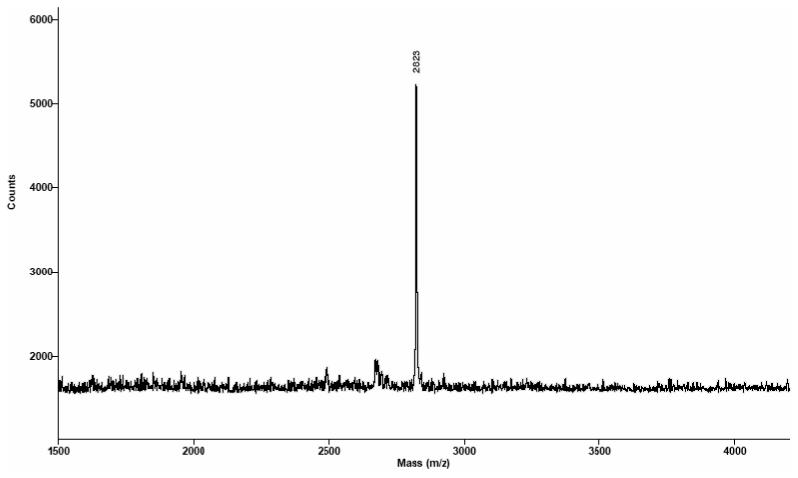
MALDI-TOF MS analysis of the DMTr-off Se-DNA (ATGGTSeGCTC). Molecular formula: C89H114N32O54P8Se; FW: 2822.8; found [M+H]+: 2823; calculated [M+H]+: 2823.8.
Table 1.
MALDI-TOF MS data of Se-oligonucleotides
| entry m/z | Se-oligonucleotides | measured (calcd.) |
|---|---|---|
| a. | Se-DNA-1 (5′-GTSeGTACAC-3′) C79H101N30O46P7Se: FW 2502.5 |
[M+H]+: 2503 (2503.5) |
| b. | Se-DNA-2 (5′-ATGGTSeGCTC-3′) C89H114N32O54P8Se: FW 2822.8 |
[M+H]+: 2823 (2823.8) |
| c. | Se-DNA-3 (5′-GGATSeGGGCG-3′) C91H112N42O51P8Se: FW 2936.9 |
[M+H]+: 2938 (2937.9) |
| d. | Se-DNA-4 (5′-GCGTSeATACGC-3′) C98H125N38O58P9Se: FW 3120.4 |
[M+H]+: 3121 (3121.4) |
| e. | Se-RNA-1 (5′-GGTSeAUUGCGGUACC-3′) C135H169N52O97P13Se: FW 4553.7 |
[M+H]+: 4555 (4554.7) |
| f. | Se-RNA-2 (5′-UGAGCTSeUCGGCUC-3′) C124H157N45O91P12Se: FW 4184.5 |
[M+H]+: 4186 (4185.5) |
| g. | Se-RNA-3 (5′-CUGUGTSeUCGAUCCACAG-3′) C162H204N60O118P16Se: FW 5454.2 |
[M+H]+: 5455 (5455.2) |
To compare the thermostability of the Se-oligonucleotides with their native ones, the UV melting experiment was carried out. The samples (2 μM DNA or RNA duplexes or hairpin loop), disolved in the buffer of 50 mM NaCl, 10 mM NaH2PO4-Na2HPO4 (pH 6.5), 0.1 mM EDTA and 10 mM MgCl2, were heated to 85 °C and allowed to cool down to room temperature slowly. The oligonucleotide samples were then stored at 5 °C for two hours before the UV melting measurement were carried out. All of the denaturing curves were collected by Cary 300 UV-Visible Spectrophotometer with a temperature controller at a heating rate of 0.5 °C/min. The data were analyzed in accordance with the convention of Puglisi and Tinoco.20 The melting temperatures and typical curves are presented in Table 2 and the Supporting Materials, respectively. There were no significant differences between the melting temperatures of the 2′-Se-oligonucleotide duplexes and the native ones. In addition, the Se-modifid and native tRNA TΨC loops have almost the same melting temperatures. Therefore, our UV thermostability study indicates that this selenium modification does not significantly alter the duplex and loop structures, which is consistent with our previous UV melting results of the 2′-Se-DNA and RNA oligonucleotides.11a,c As the UV melting study can not provide detailed information on the structure change, NMR structural analysis will be needed for the further characterization.
Table 2.
UV Melting temperatures of the 2′-Se-RNAs & -DNAs
| entry | DNA/RNA pairs | melting temp. (°C, native) |
|---|---|---|
| RNA | ||
| a. | 5′-GGTSeAUUGCGGUACC-3′ 3′-CCA--UAACGCCAUGG-5′ |
53.5 (54.2) |
| b. | 5′-UGAGCTSeUCGGCUC-3′ 3′-ACUCGA--AGCCGAG-5′ |
47.2 (47.8) |
| c. | 5′-CUGUGTSeUCGAUCCACAG-3′ (the tRNA TΨC hairpin loop) |
51.7 (52.4) |
| DNA | ||
| d. | DNA 5′-ATGGTSeGCTC-3′ 3′-TACCA--CGAG-5′ |
42.9 (42.6) |
| e. | 5′-GGATSeGGGCG-3′ 3′-CCTA--CCCGC-5′ |
40.1 (40.2) |
In our crystallization study, we found that the Se-DNA (GTSeGTACAC, self-complementary) crystallized in two to three weeks in the native buffer, where the native DNA (GTGTACAC) crystallized over two months.21,22 When the Nucleic Acid Mini Screen kit (Hampton Research, with 24 diversified crystallization buffers) was screened, to our pleasant surprise, we found that the Se-DNA crystallized overnight in 20 out of 24 buffers (buffer #5-24). In contrast, the native DNA did not crystallize at all over many weeks in these Hampton buffers. It was also reported15b that the two RNAs (12mer and 16mer) containing the 2′-Se-guanosines crystallized respectively in 17 and 33 buffers, while their corresponding native RNAs crystallized in 15 and 24 buffers, individually. Though the crystallization conditions of these Se-RNAs expanded slightly, it appears that these two Se-RNAs, which crystallized mostly as thin needle crystals, behaved similarly as many Se-Met-derivatized proteins that required the fine-tuning of their native crystallization conditions. By contrast, the majority of these Se-DNA crystals grown under the new buffers were larger than the native DNA crystals grown under the native buffer conditions, and these Se-DNA crystals also diffracted well. Our results reveal that the 2′-Se derivatization of thymidine facilitated the crystal packing and growth, and appeard to help expand the crystallization conditions.
Finally, the structure of the Se-DNA crystal, grown under the buffer #7, was determined at 1.40 Å resolution via the Se SAD phasing and the refinement. We found that the determined Se-DNA structure (Figure 3) is superimposable to the native structure (2.0 Å resolution) with the same space group,21 indicating that this Se derivatization does not alter the native structure significantly. In addition, this higher structure resolution allows observation of ordered 43-water molecules and other structure features. The Se-derivatized structure reveals that the 2′-Se-furanose displays the 3′-endo sugar pucker, which is consistent with the sugar pucker of A-form DNAs and RNAs, and the 2′-methylseleno group is placed in the minor groove of the duplex. Data collection, phasing and refinement statistics of the determined Se-DNA structure (2HC7) are listed in the Supporting Materials.
Figure 3.
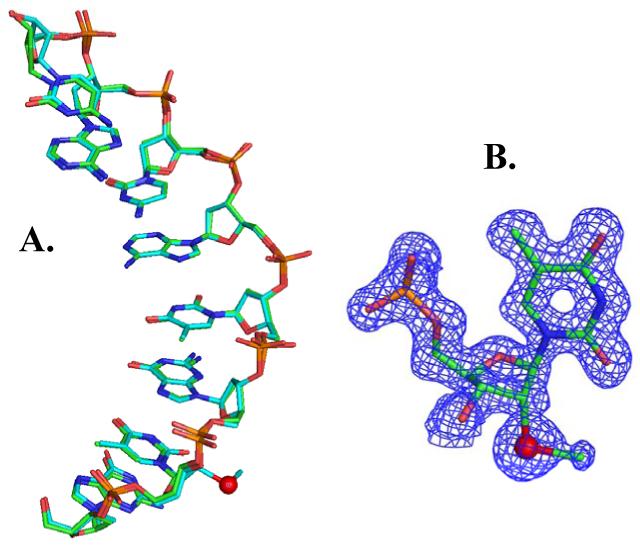
DNA crystal structures (Se in red). (A) Superimposed structure comparison of the native DNA (GTGTACAC, 1DNS in green) and the Se-derivatized DNA (GTSeGTACAC, 2HC7 in cyan). (B) The structure model and the electron density map of 2′-Se-ribothymidine in the structure.
In conclusion, we have successfully synthesized the 2′-Se-thymidine phosphoramidite (5) as an analog of both thymidine and ribothymidine phosphoramidites for the solid-phase Se-oligonucleotide synthesis. Subsequently, we have incorporated it into DNAs and RNAs with over 99% coupling yield. Our thermal denaturing study of the Se-modified DNAs and RNAs also reveals that this Se-derivatization did not significantly change the stability of the structures of the DNA and RNA duplexs and the tRNA TΨC loop. Furthermore, our X-ray crystal structure study with a Se-modified DNA octamer (GTSeGTACAC) shows that the Se-derivatized DNA structure (1.40 Å resolution) is virtually identical to the native structure (2.0 Å resolution) with the same space group. More importantly, we observed that this Se modification dramatically expanded the crystallization conditions of the Se-DNA and largely facilitated crystal growth while retaining the high diffraction quality. Besides for DNA investigations, this 2′-Se-thymidine can also serve as a ribothymidine analog for the structure and function studies of tRNAs and rRNAs. As the Se derivatization can facilitate the phase determination and crystallization, it will significantly impact the structure and function studies of nucleic acids and their protein complexes using X-ray crystallography. In addition, we are in a process of further investigating the mechanism of the crystal growth facilitated by the Se derivatization.
Supplementary Material
Scheme 1.
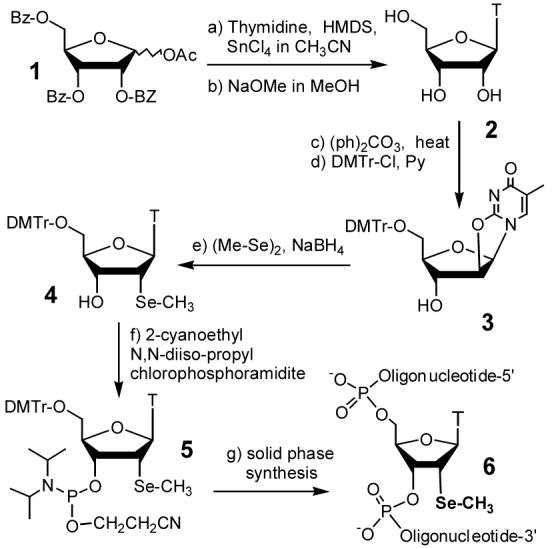
Synthesis of 5′-DMTr-2′-MeSe-thymidine phosphoramidite (5) and oligonucleotides containing this Se derivatization (6).
Acknowledgment
We thank Dr. Anand Saxena at NSLS beamline X12C for his help in the data collection, and thank Dr. Abdalla Hassan at Georgia State University for his comments and discussion. This work was partially supported by the GSU Research Program, the NIH (GM069703), and the NSF (MCB-0517092).
References
- (1).Nougayrède J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Science. 2006;313:848. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- (2).Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Nature. 2004;430:45. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- (3).Benoff B, Yang H, Lawson C, Parkinson G, Liu J, Blatter E, Ebright YW, Berman HM, Ebright RH. Science. 2002;297:1562. doi: 10.1126/science.1076376. [DOI] [PubMed] [Google Scholar]
- (4).Watson JD, Crick GHC. Nature. 1953;171:737. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- (5).Lima LMTR, Cordeiro Y, Tinoco LW, Marques AF, Oliveira CLP, Sampath S, Kodali R, Choi G, Foguel D, Torriani I, Caughey B, Silva JL. Biochemistry. 2006;45:9180. doi: 10.1021/bi060532d. [DOI] [PubMed] [Google Scholar]
- (6)(a).Ferre-D’Amare AR, Zhou K, Doudna JA. Nature. 1998;395:567. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]; (b) Egli M. Curr. Opin. Chem. Biol. 2004;8:580. doi: 10.1016/j.cbpa.2004.09.004. [DOI] [PubMed] [Google Scholar]; (c) Holbrook SR, Kim SH. Biopolymers. 1997;44:3. doi: 10.1002/(SICI)1097-0282(1997)44:1<3::AID-BIP2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (7)(a).Yang W, Hendrickson WA, Crouch RJ, Satow Y. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]; (b) Hendrickson WA, Ogata CM. Methods Enzymol. 1997;276:494. doi: 10.1016/S0076-6879(97)76074-9. [DOI] [PubMed] [Google Scholar]; (c) Liu D, Lepore BW, Petsko GA, Thomas PW, Stone EM, Fast W, Ringe D. Proc. Natl. Acad. Sci. USA. 2005;102:11882. doi: 10.1073/pnas.0505255102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Carrasco N, Ginsburg D, Du Q, Huang Z. Nucleosides Nucleotides Nucleic Acids. 2001;20:1723. doi: 10.1081/NCN-100105907. [DOI] [PubMed] [Google Scholar]
- (9).Du Q, Carrasco N, Teplova M, Wilds CJ, Egli M, Huang Z. J. Am. Chem. Soc. 2002;124:24. doi: 10.1021/ja0171097. [DOI] [PubMed] [Google Scholar]
- (10)(a).Teplova M, Wilds CJ, Wawrzak Z, Tereshko V, Du Q, Carrasco N, Huang Z, Egli M. Biochimie. 2002;84:849. doi: 10.1016/s0300-9084(02)01440-2. [DOI] [PubMed] [Google Scholar]; (b) Wilds CJ, Pattanayek R, Pan C, Wawrzak Z, Egli M. J. Am. Chem. Soc. 2002;124:14910. doi: 10.1021/ja021058b. [DOI] [PubMed] [Google Scholar]
- (11)(a).Buzin Y, Carrasco N, Huang Z. Org. Lett. 2004;6:1099. doi: 10.1021/ol0365077. [DOI] [PubMed] [Google Scholar]; (b) Carrasco N, Huang Z. J. Am. Chem. Soc. 2004;126:448. doi: 10.1021/ja0383221. [DOI] [PubMed] [Google Scholar]; (c) Carrasco N, Buzin Y, Tyson E, Halpert E, Huang Z. Nucleic Acids Res. 2004;32:1638. doi: 10.1093/nar/gkh325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Salon J, Chen GX, Portilla Y, Germann MW, Huang Z. Org. Lett. 2005;7:5645. doi: 10.1021/ol052270y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Logan G, Igunbor C, Chen G-X, Davis H, Simon A, Salon J, Huang Z. Synlett. 2006;10:1554. [Google Scholar]
- (12).Carrasco N, Williams JC, Brandt G, Wang S, Huang Z. Angew. Chem. Int. Ed. 2006;45:94. doi: 10.1002/anie.200502215. [DOI] [PubMed] [Google Scholar]
- (13).Brandt G, Carrasco N, Huang Z. Biochemistry. 2006;45:8972. doi: 10.1021/bi060455m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14)(a).Hobartner C, Micura R. J. Am. Chem. Soc. 2004;126:1141. doi: 10.1021/ja038481k. [DOI] [PubMed] [Google Scholar]; (b) Hobartner C, Rieder R, Kreutz C, Puffer B, Lang K, Polonskaia A, Serganov A, Micura R. J. Am. Chem. Soc. 2005;127:12035. doi: 10.1021/ja051694k. [DOI] [PubMed] [Google Scholar]
- (15)(a).Serganov A, Keiper S, Malinina L, Tereshko V, Skripkin E, Hobartner C, Polonskaia A, Phan AT, Wombacher R, Micura R, Dauter Z, Jaschke A, Patel DJ. Nat. Struct. Mol. Biol. 2005;12:218. doi: 10.1038/nsmb906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moroder H, Kreutz C, Lang K, Serganov A, Micura R. J. Am. Chem. Soc. 2006;128:9909. doi: 10.1021/ja0621400. [DOI] [PubMed] [Google Scholar]
- (16).Egli M, Pallan PS, Pattanayek R, Wilds CJ, Lubini P, Minasov G, Dobler M, Leumann CJ, Eschenmoser A. J. Am. Chem. Soc. 2006;128:10847. doi: 10.1021/ja062548x. [DOI] [PubMed] [Google Scholar]
- (17)(a).Becker HF, Motorin Y, Florentz C, Giege R, Grosjean H. Nucleic Acids Res. 1998;26:3991. doi: 10.1093/nar/26.17.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sprinzl M, Vassilenko KS. Nucleic Acid Res. 2005;33:D139. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McCloskey JA, Rozenski J. Nucleic Acids Res. 2005;33:D135. doi: 10.1093/nar/gki015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18)(a).Vorbruggen H, Bennua B. Chem. Ber. 1981;114:1279. [Google Scholar]; (b) Huang Z, Benner SA. J. Org. Chem. 2002;67:3996. doi: 10.1021/jo0003910. [DOI] [PubMed] [Google Scholar]
- (19)(a).Mansuri MM, Starrett JE, Wos JA, Tortolani DR, Brodfuehrer PR, Howell HG, Martin JC. J. Org. Chem. 1989;54:4780. [Google Scholar]; (b) Saito Y, Zevaco TA, Agrofoglio LA. Tetrahedron. 2002;58:9593. [Google Scholar]; (c) Shi J, Du J, Ma T, Pankiewicz KW, Patterson SE, Tharnish PM, McBrayer TR, Stuyver LJ, Otto MJ, Chu CK, Schinazi RF, Watanabe KA. Bioorg. Med. Chem. 2005;13:1641. doi: 10.1016/j.bmc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- (20).Puglisi JD, Tinoco I., Jr. Methods Enzymol. 1989;180:304. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- (21).Jain S, Zon G, Sundaralingam M. Biochemistry. 1989;28:2360. doi: 10.1021/bi00432a002. [DOI] [PubMed] [Google Scholar]
- (22).Thota N, Li XH, Bingman C, Sudaralingam M. Acta Cryst. 1993;D49:282. doi: 10.1107/S0907444992007522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.