

Abstract
Copper cluster sites in biology exhibit unique spectroscopic features reflecting exchange coupling between oxidized Cu’s and e− delocalization in mixed valent sites. These novel electronic structures play critical roles in O2 binding and activation for electrophilic aromatic attack and H atom abstraction, the 4e−/4H+ reduction of O2 to H2O, and in the 2e−/2H+ reduction of N2O. These electronic structure/reactivity correlations are summarized below.
Cu proteins play central roles in Fe, Cu, and O2 metabolism, are related to a range of genetic diseases and are important in biotechnology, detoxification, and the elimination of greenhouse gases. Understanding Cu biochemistry on a molecular level provides mechanisms to improve or inhibit these processes and enhance drug design. The Cu proteins involved in O2 binding, activation, reduction to H2O and the reduction of N2O to water and dinitrogen are summarized in Figure 1. The term “coupled” is used here to refer to the antiferromagnetic (AF) “coupling” between paramagnetic metal centers that can lead to a diamagnetic Stot=0 ground state. If two Cu(II)’s, S=1/2, directly overlap they will spin pair. If, however, they are far enough apart so that their d orbitals do not directly overlap but have a bridging ligand this can provide a superexchange pathway (i.e. a delocalized molecular orbital) between the two paramagnetic Cu(II)’s that results in their spin pairing, indirectly through overlap with the bridge. This is described by the exchange Hamiltonian H =−2JSA·SB which spin couples the two S=1/2’s on CuA and CuB to form total spins Stot=1 and 0 where for AF coupling the Stot=0 is lower in energy by 2J (J<0).
Figure 1.

Multinuclear Cu Sites in Biology
In this paper we will: 1) consider the unique spectral features of the coupled binuclear Cu proteins, hemocyanin (Hc), catechol oxidase, and tyrosinase (Ty), that reflect a novel electronic structure that allows their reversible binding of O2 (a spin forbidden process) and its activation for electrophilic attack on an aromatic substrate by Ty; 2) contrast this electronic structure to that of the non-coupled binuclear Cu enzymes (i.e. no magnetic interaction between the two Cu(II)’s S=1/2) to evaluate the contribution of these differences in AF exchange coupling to the reaction mechanisms, where the non-coupled binuclear Cu sites in dopamine α-monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM) activate O2 for H-atom abstraction; 3) Extend these studies to the trinuclear Cu cluster site in the multicopper oxidases, where the exchange coupling among the three coppers plays a central role in the 4e−/4H+ reduction of O2 to H2O; and 4) Consider how the interactions among the coppers in the μ4 sulfide bridged tetranuclear Cuz cluster promote the 2e−/2H+ cleavage of the N-O bond by N2O reductase.
1. Coupled Binuclear Copper Proteins
From spectroscopy and crystallography, Hc (reversible O2 binding), catechol oxidase (O2 binding and catechol oxidation to quinone) and Ty (O2 binding, oxidation and monooxygenation of phenol to catechol) all have equivalent geometries (μ-η2:η2 Cu(II)2O2, side-on peroxide bridged binuclear Cu(II) sites coordinated to the protein by 3 His ligands on each Cu) and electronic structures (vide infra).1 Their range of functions increases from Hc to Ty, which has been attributed to differences in substrate accessibility to their coupled binuclear Cu sites.
1.1 Unique Spectroscopic Features → Novel Electronic Structure
To understand the unique spectral features of the oxygenated sites of these proteins we first consider what is normal for a Cu(II)-peroxide bond. Cu(II) is d9 and thus has one half occupied valence d-orbital, either dx2−y2 for tetragonal or dz2 for trigonal bipyramidal geometries. Peroxide has a doubly degenerate highest occupied molecular orbital (HOMO) set which will split in energy upon binding to Cu(II), the π*σ being stabilized to deeper binding energy due to σ bonding to the half occupied d orbital and the π*v (vertical) not being very affected by bonding as it is perpendicular to the Cu(II)-(O22−)plane (Figure 2A).2
Figure 2.

Electronic structure of (A) End-on Cu-O22− complex (B) End-on bridged [Cu2-O22−] complex and (C) Side-on bridged OxyHc.
In a peroxide bridged binuclear Cu(II) complex as in the [(TMPA)Cu]2O2 dimer (trigonal bipyramidal at each Cu(II)), one further has to take symmetric and antisymmetric combinations of the half occupied dz2 orbitals on the Cu’s and then allow for bonding with the occupied peroxide π*σ valence orbitals.3 As shown in Figure 2B only one contribution of dz2 orbitals has net σ overlap with the peroxide π*σ orbital causing stabilization of the π*σ orbital and a destabilization of the dz2 A + dz2 B molecular orbital. This leads to spin pairing of the two electrons in the dz2 orbitals of the two coppers and the antiferromagnetically coupled S=0, singlet ground state. This also produces a peroxide π*σ → Cu (dz2 A + dz2 B) charge transfer (CT) transition as shown in Figure 3A. Resonance Raman (rR) excitation into this CT transition produces an O-O stretching vibration at ~830 cm−1 (Figure 3B) which is characteristic of peroxide bridging in a “normal” binuclear Cu(II) complex.3
Figure 3.
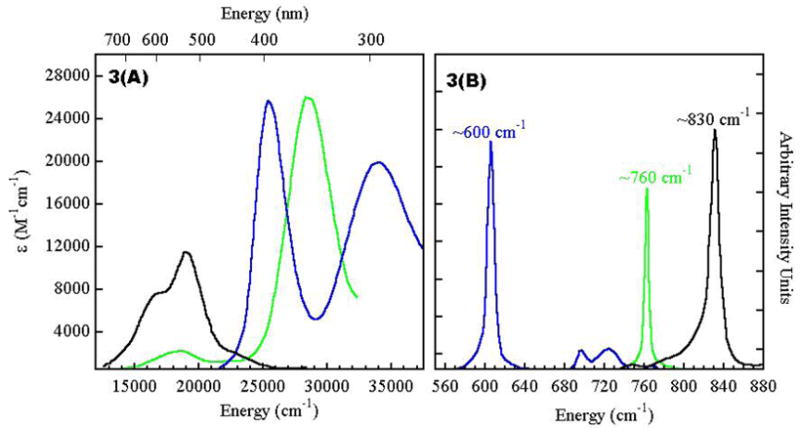
(A) Absorption and (B) Resonance Raman spectra of {[(TMPA)Cu]2O2}2+ (end-on) (—), Cu[HB(3,5-i-Pr2pz)3]2(O2) (side-on) (
 ) and {[LTMCHDCu]2O2} (bis-μ-oxo (
) and {[LTMCHDCu]2O2} (bis-μ-oxo (
 )
)
We now consider the unique geometric and electronic structure of oxy-Hc/Oxy-Ty. Again, we take the symmetric and antisymmetric combination of the dx2−y2 half occupied valence orbitals of the two coppers (here the ligand field of each Cu is square pyramidal) and allow for their bonding interactions with the peroxide π* valence orbitals, but in the side-on peroxide bridged structure of the oxy-Hc site (Figure 2C). Again the π*σ orbital is stabilized and the Cu dx2−y2A + dx2−y2 B is destabilized due to σ bonding. However, in the side-on bridged structure there are two σ-bonding interactions of the peroxide with each copper and this leads to a very large bonding-antibonding interaction.4 Thus, a strongly AF stabilized singlet ground state and an intense O22− π*σ → Cu(dx2−y2A + dx2−y2 B) CT transition, which is shifted to higher energy, are observed (Figure 3A, green vs. black). Importantly, rR excitation into this CT transition shows a very low O-O stretching frequency (νO-O) at ~750 cm−1 (Figure 3B). The large peroxide σ donation of electron density from the π*σ orbital (which is antibonding with respect to the O-O bond) to the Cu(II)’s should increase not decrease the strength of the O-O bond. However, there is an additional bonding interaction that occurs in the side-on peroxide bridged structure. The LUMO on the peroxide is the σ* (Figure 2C) which bonds with the occupied dx2−y2 A + dx2−y2 B combination of d-orbitals on the Cu’s which is the HOMO. This back-bonding shifts some of the electron density into the O22− σ* orbital which is strongly antibonding with respect to the O-O bond and leads to the very low νO-O.4
In summary, the extremely covalent σ bonding between the two Cu(II)’s through the μ-η2:η2 peroxide π*σ orbital leads to the AF coupled singlet ground state and the backbonding of electron density from the Cu (dx2−y2A + dx2−y2 B) HOMO into the peroxide σ* LUMO leads to an extremely weak O-O bond activated for cleavage.
1.2 Reaction Coordinate for O2 Binding
DeoxyHc/Ty with two d10 Cu(I) centers binds triplet O2 reversibly to form oxyHc/oxyTy which has the AF coupled singlet ground state. Thus, this reaction is spin forbidden. To obtain a reaction coordinate for reversible O2 binding, we started with the μ-η2:η2 structure, moved the peroxide out of the molecular plane, and geometry optimized the rest of the structure.5 The structure first butterflies, then goes to a μ-η1:η2 asymmetric structure, and then to an end-on bridged structure in the reversible loss of O2 (Figure 4A). These structures maximize the metal-ligand overlaps along this reaction coordinate. As the peroxide moves away from the coppers, its negative charge decreases as does the positive charge on the coppers. Thus, electron density is being transferred from the peroxide to the Cu(II)’s. Importantly, both coppers are reduced at the same rate even in the asymmetric μ-η1:η2 structure. Thus, the reversible loss of peroxide as O2 involves simultaneous two electron transfer.
Figure 4.
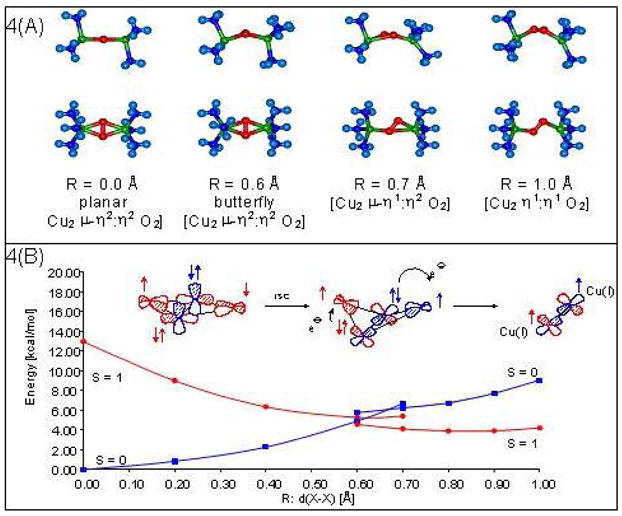
(A)The reaction coordinate of O2 binding by Hc. View along the O-O (Top) and perpendicular to initial Cu2O2 plane (bottom). (B) Potential-energy surfaces for the interconversion of OxyHc and DeoxyHc in triplet and Singlet state. R:d(X-X) is the distance between the center of the O-O and Cu-Cu vectors. R:d(X-X) <~0.6 and >~0.6 represents symmetric and non-symmetric O2 coordination, respectively.
In Figure 4B we consider how the spin changes along this reaction coordinate. On the far left is the AF stabilization of the singlet ground state of the side-on peroxide bridged structure through its π*σ orbital. However, as the peroxide moves out of the molecular plane to the butterflyed structure, the singlet/triplet splitting collapses and the triplet is in fact slightly lower in energy. This is because the spin on each Cu covalently delocalizes into a different peroxide σ* orbital. These are close to orthogonal in the butterflyed structure which favors the triplet ground state. From here, one electron of the same spin can be transferred from each σ* orbital to each Cu leading to loss of O2 in its triplet 3Σg− ground state.5
Thus, the stabilization of the triplet ground state of O2 is lost by charge transfer from the remote Cu’s and the singlet structure is then stabilized by the formation of an efficient superexchange pathway (the π*σ orbital overlap) along the reaction coordinate.
1.3 Reaction Coordinate of Monooxygenation
In early literature we found that oxyTy had the same geometric and electronic structure as oxyHc but that it differed for Hc in having substrate access and coordination directly to the copper in a trigonal bipyramidal distorted structure. This led to the generally accepted monooxygenation mechanism for oxyTy shown in Figure 5.1,6,7 Phenolate substrate binds directly to the side-on peroxy bridged oxyTy site (oxy-T). The trigonal pyramidal distortion leads to electrophilic attack and hydroxylation at the ortho-position to produce a bound catecholate (met-D). Two electron oxidation leads to the corresponding quinone product and a reduced (deoxy) site capable of O2 binding for further turnover. The interesting issue now is whether the side-on oxyTy structure directly reacts with the aromatic ring or whether phenolate coordination leads to a bis-μ oxo structure (vide infra) and this does the electrophilic attack on the ring.
Figure 5.

Molecular mechanism of Ty catalysis. The two possible structures of substrate bound oxy-T are expanded.
The latter possibility was raised by the results of Tolman et al. who showed that the side-on peroxy bridged structure could convert to the bis-μ oxo structure with certain chelating ligands (Figure 6A).8 Cu K pre-edge X-ray absorption data showed that conversion of the side-on to the bis-μ oxo isomer leads to an increase in energy of the Cu 1s→3d transition by 1.9 eV indicating that the Cu(II) is oxidized to Cu(III) in the bis-μ oxo structure.9 The orbital correlation diagram for this interconversion is given in Figure 6B. Starting from the side-on bridged structure on the left, the O-O bond elongates from 1.4 Å to 2.3 Å in the bis-μ oxo structure (the Cu-O distance goes from 1.92 to 1.81Å). The O22− σ* orbital thus drops in energy to below the HOMO on the coppers resulting in oxidation to two Cu(III)’s and reduction of the peroxide to the bridged oxide level.10 This produces a low energy intense μ-O2− → Cu (dx2−y2A − dx2−y2 B) LUMO CT transition (Figure 3A, blue), and rR excitation into this transition now shows an intense vibrational peak at 600 cm−1 corresponding to the Cu2(O)2 symmetric stretch (Figure 3B). Thus, we now have two LUMO’s that are frontier molecular orbitals (FMO) capable of electrophilic attack on the occupied π orbitals of the aromatic ring (Figure 6C). In the side-on bridged structure, there is a Cu LUMO with significant peroxide π*σ character due to the strong donor interaction of the peroxide with the Cu. In the bis-μ oxo structure, the Cu based LUMO now has significant σ* character due to the strong oxide σ donor interaction.
Figure 6.
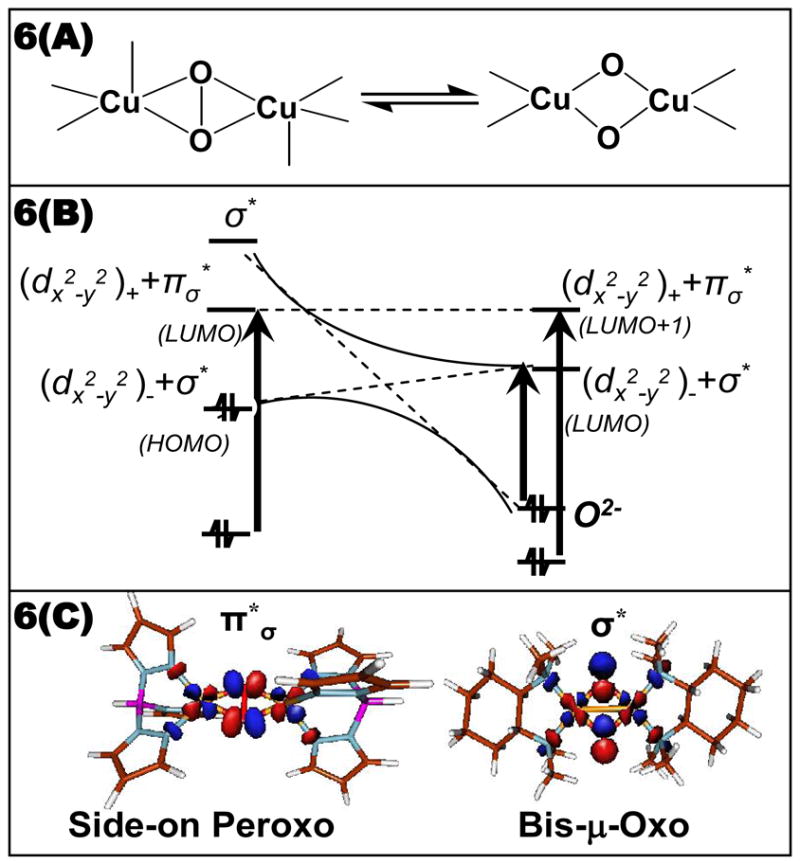
(A) Side-on peroxo (left), Bis-μ-Oxo (right) correlation (B) electronic structure correlation (C) FMO’s (ie. LUMO’s).
From model studies, both the π* LUMO of the side-on peroxide and the σ* LUMO of the bis-μ oxo structures are capable of electrophilic reactions with aromatic substrates. From Figure 7A, in the Karlin bidentate chelate ligand system11, an O2 intermediate is trapped at low temperature with an electron withdrawing nitro substituent on the bridging ring. From rR data (Figure 7B), this species shows a ~750 cm−1 vibration characteristic of the side-on peroxo bridged species with no indication of a 600 cm−1 feature characteristic of the bis-μ oxo species (upper limit of < 0.1%).12 As shown in Figures 7B and 7C, the ~750 cm−1 feature decreases as the 1320 cm−1 feature, characteristic of the C-O stretch of the hydroxylated phenolate product, increases.
Figure 7.
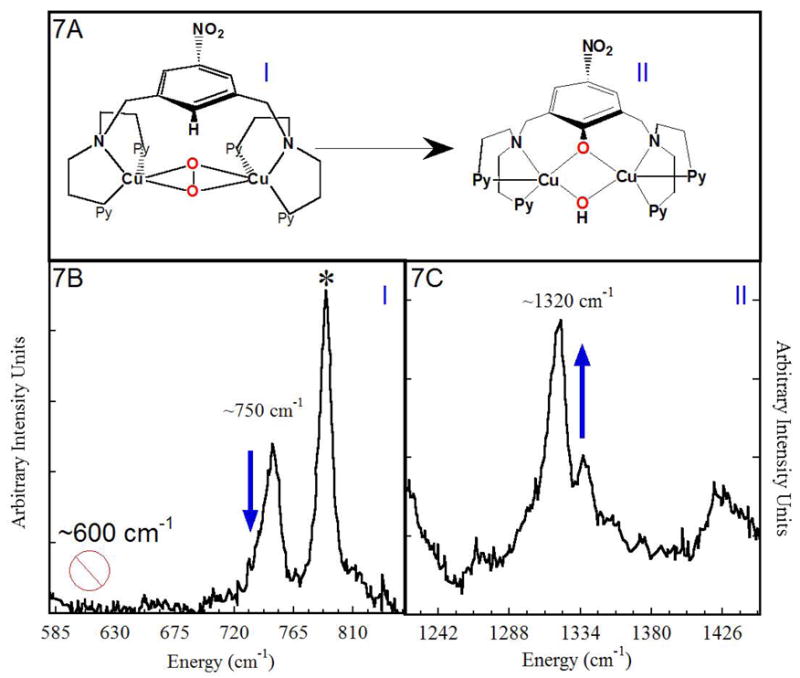
(A) Conversion of the side-on peroxide intermediate [Cu2(NO2-XYL)(O2)]2+ to [CuII2(NO2-XYL-O−)(OH)]2+. (B) & (C) Change in rR with time: loss of side-on peroxo stretch correlates with increase of C-O stretch.
From Figure 8, the Stack diamine ligated complex binds O2 as the side-on peroxo bridged species.13 Addition of exogenous phenolate leads to the loss of the ~750 cm−1 νO-O vibration and appearance of the 600 cm−1 feature characteristic of the bis-μ oxo species. Thus, coordination of the phenolate to the Cu likely through a trigonal bipyramidal rearrangement into the equatorial plane, converts the side-on peroxide to the bis-μ oxo species. This goes on to hydroxylate the phenolate to a mixture of catecholate and quinone products. Thus, both the side-on peroxo (through a π* electrophilic mechanism) and the bis-μ oxo (through a σ* electrophilic mechanism) binuclear Cu sites can hydroxylate aromatic substrates.
Figure 8.
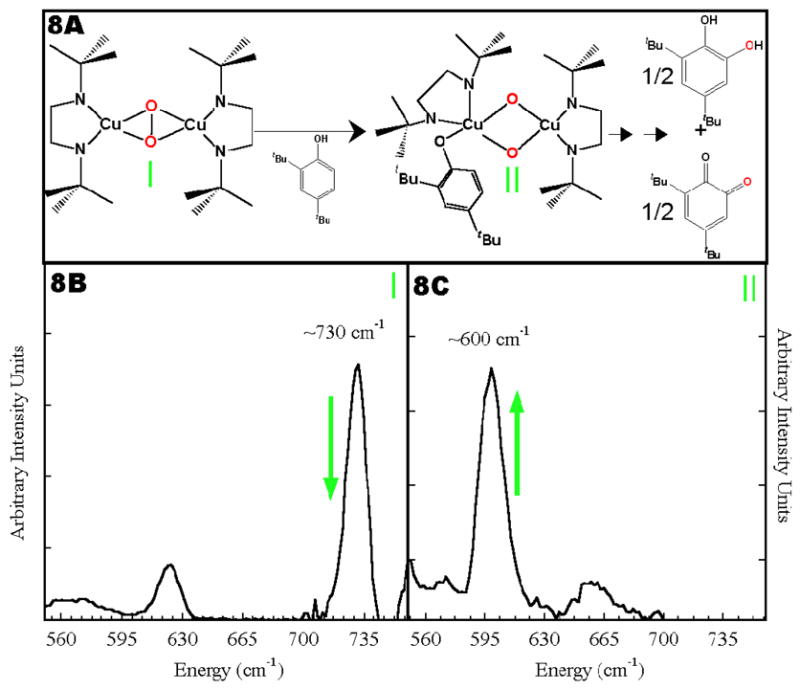
(A) Reaction of Side-on peroxo {[(DBED)Cu]2O2}2+.(B) & (C) Change in rR with (tBu)2Ph(O)− coordination.
2. Non-coupled Binuclear Cu Enzymes
PHM and DβM, involved in peptidic hormone production and the control of neurotransmitters, both catalyze substrate C-H bond hydroxylation by H-atom abstraction.14 The active sites in these enzymes are non-coupled in that they have two Cu(II)’s which each show spectroscopic features indicative of an isolated Cu(II) S=1/2 center. This is consistent with the crystallography on PHM which shows that the two Cu(II)’s are 11Å apart with no bridging ligation (only H2Os in the interdomain cavity between the Cu’s).15 Reasonable descriptions of the geometric and electronic structure of each Cu(II) center have been obtained through a combination of crystallography, EXAFS, MCD spectroscopy and DFT calculations.16,17 As shown in Figure 9 CuM is the catalytic center which has an axial Met, two equatorial His and two equatorial H2O/OH− sites for O2 reactivity. CuH has 3 His and a water ligand in a D2d distorted tetragonal geometry and supplies the extra e− required for catalysis to CuM over a distance of 11Å. There has been much discussion as to how this ET might take place.18
Figure 9.
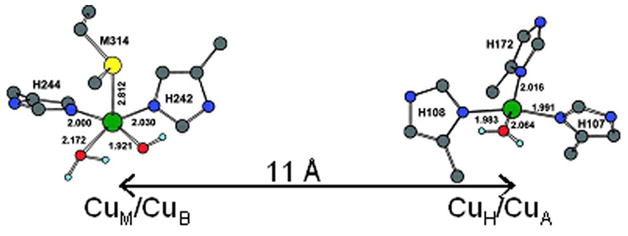
Geometry-optimized structures of the resting oxidized CuM and CuH sites in PHM.
Thus the reaction of the non-coupled binuclear Cu enzymes requires O2 activation by a single Cu center, which until recently was thought to involve a CuM(II)-OOH species.14 However from spectroscopic and electronic structure studies on a Cu(II)-hydroperoxide model complex, this species is not activated for H-atom abstraction.19 From Figure 10A the FMO only has 2% character on the distal oxygen for electrophilic attack and from rR studies on the model the νO-O is 843 cm−1 reflecting a strong O-O bond not activated for cleavage. This led us to studies of the alternative possibility of a 1e− reduction of O2 to generate a bound superoxo-CuM(II) intermediate. From the rR data in Figure 10B the νO-O of an η2 O2Cu model complex is 1043 cm−1 which is characteristic of a Cu(II)-O2− species; from its FMO which has >60% O character this species is strongly activated for electrophilic attack on H-C bonds.20
Figure 10.
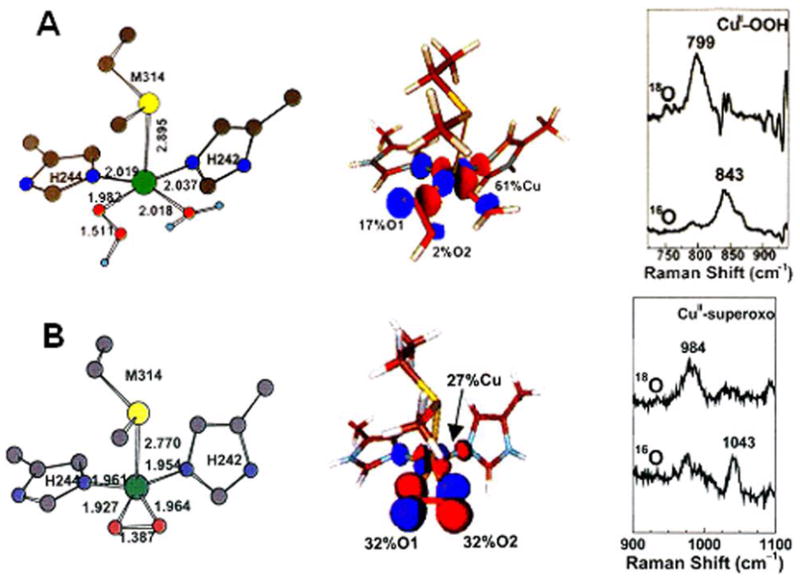
Electronic structure of the CuIIM-OOH (A) and CuIIM-superoxo (B) species. Geometry-optimized structure (left), acceptor FMO (LUMO) (middle), rR spectra in νo-o region (right).
These predictions from model studies were strongly supported by electronic structure calculations of this reaction coordinate.21 H atom abstraction by the 2e− reduced CuM(II)-hydroperoxo species (Figure 11 blue) is endergonic and in particular has an activation barrier of 37 kcal/mol. Alternatively H atom abstraction by the 1e− reduced Cu-superoxo species (Figure 11 red) is thermoneutral and has an activation barrier of only 14 kcal/mol, which is consistent with the FMO predictions. This would generate a CuM(II)-OOH and substrate radical species, which would readily react via direct OH transfer from the hydroperoxide to generate the hydroxylated product and a CuM(II)-O.− (i.e. cupric-oxyl) species. This high energy species would drive a proton coupled ET from CuH to complete the reaction cycle.
Figure 11.
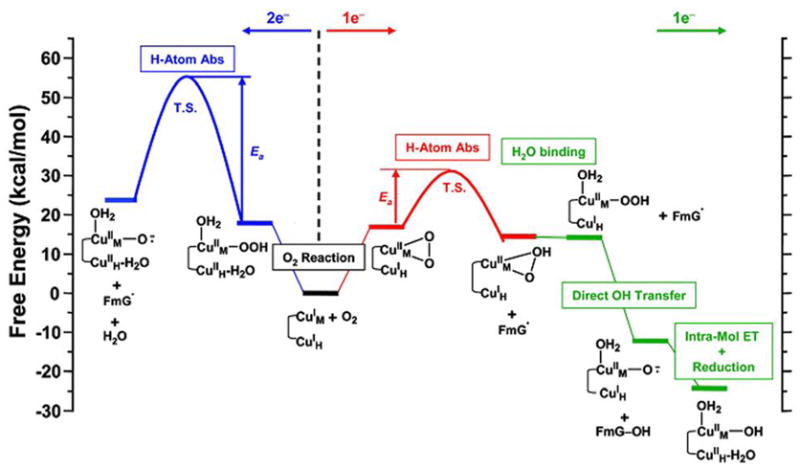
Summary of the 2e− (blue) and 1e− (red, green) reaction coordinates for the non-coupled binuclear Cu enzymes.
A comparison of the reaction coordinates for the coupled binuclear Cu enzymes (Figure 5) and non-coupled binuclear Cu enzymes (Figure 11) shows an extremely important role of the differences in the AF exchange coupling (J) in the reaction mechanism. Rapid ET requires a large electronic coupling between the donor and acceptor (HDA) which in turn is related to J∝(HDA)2.21 Thus large AF coupling leads to rapid ET. This is the case for the coupled binuclear Cu enzymes where the large value of J between the Cu’s via the bridging ligand leads to the 2e− reduction of O2 to form a Cu2O2 species capable of electrophilic aromatic attack. Alternatively in these non-coupled binuclear Cu enzymes J is very small as CuM and CuH are separated by an 11Å solvent-filled cleft. The reduction appears to proceed at one Cu via a 1e− reduced CuM(II)-O2− species which is capable of H atom abstraction. Such a CuMO2 species has been observed in the crystal structure of PHM22 and defined by rR in a model complex.23 At a later stage of the reaction a high energy species is produced (the CuM(II)-oxyl) which provides the large driving force required for ET from CuH with its low J (therefore HDA) with CuM.
3. Trinuclear Cu Cluster in the Multicopper Oxidases (MCOs)
The MCOs couple four 1e− oxidations of substrates to the 4e−/4H+ reduction of O2 to H2O.1 These can be divided into two classes: one as represented by laccase uses organic substrates which weakly to strongly interact with the protein near the type 1 (T1) Cu (vide infra) and the second represented by Fet3p has specific metal ion substrate binding sites near the T1 which tune the metal ion potential and provide ET pathways to the T1.24 The minimum structure of a MCO active site is shown in Figure 12A.
Figure 12.
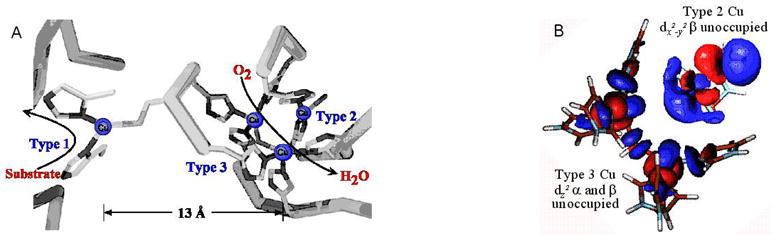
(A) Active site of the MCO’s (AO is shown here) (B) electronic structure of the TNC.
The T1 is a blue copper center capable of rapid ET through a Cys-His pathway over 13 Å to the trinuclear Cu cluster (TNC) where O2 is reduced to H2O. The TNC is comprised of a type 3 (T3) copper pair, where each Cu has three His ligands and the pair is strongly antiferromagnetically coupled (i.e. with a singlet ground state) through an OH− bridge, and a type 2 (T2) center within 3.5 Å of the T3 Cu’s having two His and a OH− ligand external to the cluster.25 The T2 Cu is not bridged to the T3 Cu’s and shows a normal Cu(II) S=1/2 EPR signal.
The electronic structure of the resting trinuclear Cu(II) cluster is given in Figure 12B. This shows that all three Cu’s have open coordination positions oriented inside the cluster. The coordination unsaturation of this highly positively charged cluster results from charged carboxylate residues within 8 Å of the cluster which destabilize H2O/OH−/O2− binding in the center of the cluster and thus tune its redox properties for O2 reduction.26
O2 intermediates were trapped to define the mechanism of O2 reduction to H2O by the MCOs. Initially, we studied a T1 depleted (T1D) derivative in laccase where the T1 Cu was replaced by a redox inactive Hg2+.27 Reduction of the TNC and reaction with O2 led to the first intermediate. From a combination of isotope ratio mass spectrometry, CD, and LT MCD (which only probes paramagnetic centers), we determined that this was a peroxy intermediate with two coppers oxidized and AF coupled and one copper reduced.27 The AF coupling required bridging ligation and this was observed in EXAFS data on peroxy T1D, which showed two Cu’s tightly bridged at 3.4 Å. Since one Cu was reduced in this intermediate, we could not directly study its interaction with the peroxide. So we prepared the peroxide adduct (PA) of the oxidized TNC in T1D.28 PA showed the same FT-EXAFS feature at 3.4 Å indicating a similar peroxide binding mode but now all Cu’s are oxidized. On binding peroxide to the oxidized TNC all the ligand field features of the T2 and T3 Cu(II)’s are perturbed, indicating an all bridged structure for the peroxide intermediate. This is consistent with the recent crystal structure of the peroxide adduct of oxidized CotA29 and the QM/MM energy minimized structure of the peroxide intermediate.30
Reaction of the fully reduced native enzyme (i.e. 4Cu(I)) with O2 generates the native intermediate (NI). As shown in Figure 13A, NI exhibits absorption features at 365 and 318 nm as well as an absorption band at 600 nm associated with an oxidized T1 Cu.31 Therefore NI is at least one more electron reduced relative to the peroxide intermediate. Associated with this, NI exhibits an unusual EPR signal (Figure 13C) with very unusual g-values below 2.0. It is different from that of an oxidized T2 Cu(II) and broadens when generated with 17O2.32 Thus, it had been assigned as an OH. bound to a reduced T2 Cu. The most direct spectroscopic probe of reduced Cu is K-edge XAS as Cu(I) exhibits a characteristic feature at 8984 eV not present in Cu(II) complexes. It is also not present in NI.31 Therefore, NI is a fully oxidized TNC, but with an EPR signal very different from that of the fully oxidized TNC of the resting enzyme (Figure 13C), and dioxygen has been fully reduced to the H2O level.
Figure 13.
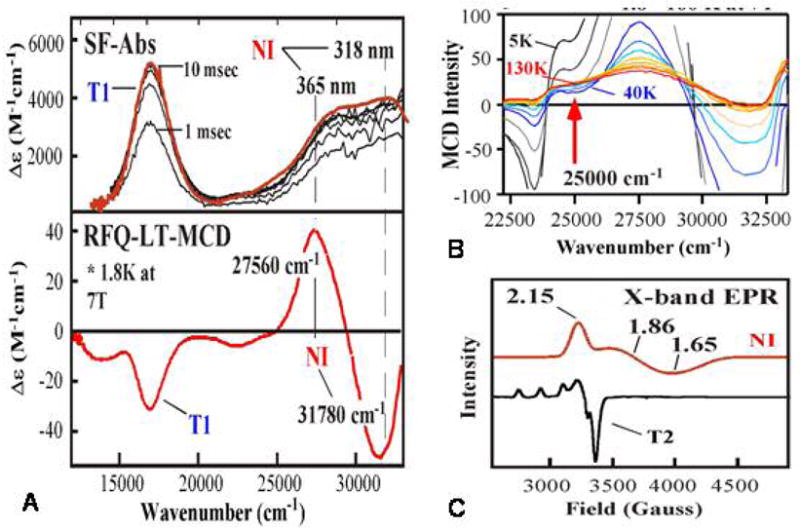
(A) Stopped-flow absorption showing the formation of NI, (B) RFQ-LT-MCD spectrum of NI, (C) VTMCD of NI, and (D) low temperature X-band EPR spectrum of NI compared to resting T2.
NI also has a characteristic derivative shaped MCD signal associated with the 365/318 nm absorption bands known as a pseudo-A term (Figure 13A).31 This has proven to be a direct probe of the geometric and electronic structure of NI. The field dependence of the MCD signal at low temperature gave a saturation magnetization curve which fit to the Brillouin function for an S = 1/2 ground state, associated with the unusual g-values. The temperature dependence of the MCD signal at a fixed high magnetic field shows a very interesting behavior in Figure 13B. Normally MCD intensity of a paramagnetic S = 1/2 center decreases as 1/T. However, the MCD intensity in Figure 13B first decreases then increases with increasing temperature indicating Boltzmann population of an excited state at 150 cm−1 with an MCD signal different from that of the S =1/2 ground state of NI.
NI has an FT-EXAFS feature indicating a Cu-Cu distance of 3.3 Å, which corresponds to a pair of Cu(II)’s of the trinuclear cluster site having a singlet/triplet splitting of ~520 cm−1. Since we are dealing with a trinuclear Cu(II) site, the singlet ground state of the pair couples with the third Cu(II) to give an Stot = 1/2 ground state; the S = 1 excited state couples with the S = 1/2 to form Stot = 1/2 and 3/2 excited states. With a single bridge, this would produce a T2 EPR signal with g-values above 2.0 and an excited state at 520 cm−1. NI has a ground state EPR signal with g-values below 2.0 and an excited state at 150 cm−1. Thus we allow for additional AF exchange interactions (i.e. bridging ligands) between additional pairs of Cu(II)’s of the TNC of NI. Allowing for a second bridge splits the excited Stot = 3/2 and 1/2 states, but does not bring the Stot = 1/2 first excited state below 440 cm−1 (~1.7J) and does not result in ground state g-values below 2.0. Addition of a third bridge now causes the S = 1/2 excited state to greatly decrease in energy (this is a result of “spin frustration” as all three S = 1/2 cannot be AF coupled in a triangle) and the ground state g-values to decrease below 2.0 (due to antisymmetric exchange in the all bridged trimer).33 Thus the experimental data on the ground state and excited state of NI require all three Cu(II)’s to be strongly exchange coupled through bridging ligands.
There were two possible structures for NI where all Cu’s are oxidized and bridged by the product of full O2 reduction: a μ3-oxo bridged structure, or a tris OH- bridged structure in which the third OH− would derive from H2O (Figure 14A top). Model complexes exist with both structures and both exhibit MCD pseudo-A terms (of opposite sign) associated with the hydroxo or oxo to Cu(II) CT transitions of the trinuclear Cu(II) cluster (Figure 14A bottom).34 The mechanism for pseudo-A terms requires two perpendicular CT transitions being spin-orbit coupled in a third direction at one center. For the tris OH− system, this would involve CT transitions from two OH− ligands to one Cu(II) center which provides the spin-orbit coupling. For the μ3-oxo structure, this involves oxo CT transitions to two Cu(II) centers which are spin-orbit coupled by the oxo bridge. From the temperature dependent MCD spectrum of NI shown in Figure 14B, the CT transitions forming the pseudo-A term involve different Cu centers which is only consistent with a μ3-oxo, all-bridged structure for NI.
Figure 14.
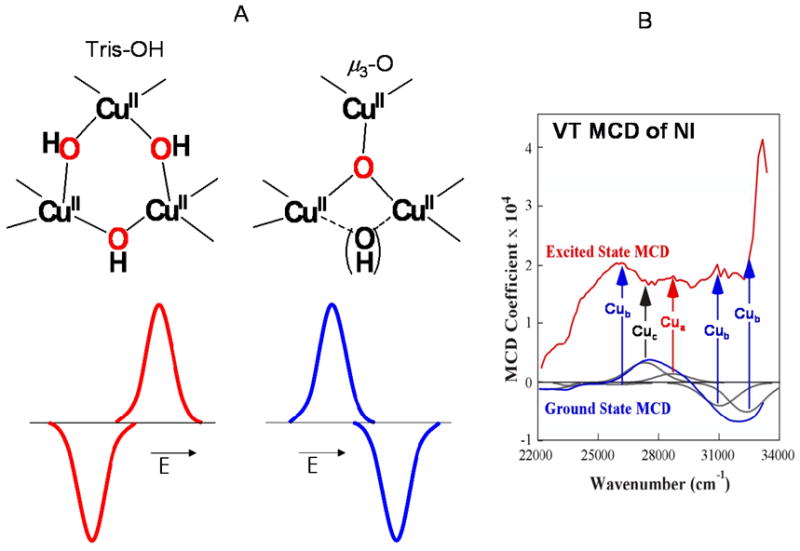
(A) Predicted and observed MCD signs for the two possible structures of NI and (B) ground and excited state MCD spectra of NI, where bands are grouped together according to their different temperature dependencies
The above spectroscopic studies of O2 intermediates in the MCO’s have led to the molecular mechanism of O2 reduction to H2O in Figure 15. The reduction of O2 by the fully reduced MCO involves two 2e− steps. From our kinetic studies the first is rate determining and the second is fast, therefore it is effectively a 4e− process.1 The second step involves the 2e− reductive cleavage of the O-O bond. It has a large driving force due to the 2e− reduction potential of peroxide and from mutagenesis studies it is proton assisted with a Glu near the T3 providing the proton for O-O bond cleavage. The NI is a fully oxidized form of the enzyme but is different from the resting form as it has an internal μ3-oxo bridge. These interconvert and the rate of decay of NI to the resting is very slow due to the reorganization of the μ3-oxo bridge (from O2) to the external position on the T2 Cu(II). This decay of NI is too slow to be in the catalytic cycle, whereas the reduction of NI is fast (>1000s−1)35 and this is the catalytically relevant fully oxidized form of the MCO’s. ET from the T1 to the TNC is fast because the μ3-oxo bridge provides an effective superexchange pathway to the T2 Cu.
Figure 15.
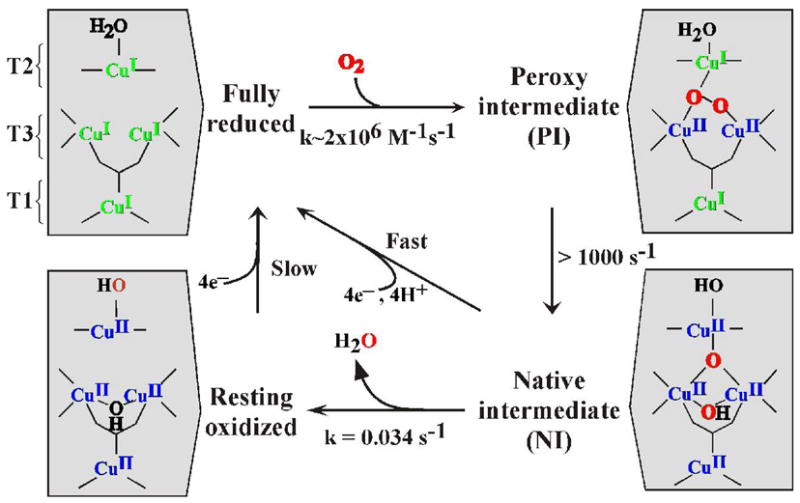
MCO mechanism
4. μ4-Sulfide bridged tetranuclear CuZ cluster in Nitrous Oxide Reductase
The tetranuclear CuZ cluster catalyzes the 2e−/2H+ cleavage of the N-O bond in N2O.36 From the crystal structure, electrons enter at the mixed valent binuclear CuA center of one subunit and are transferred over a 10 Å super-exchange pathway to the CuZ cluster of a 2nd subunit where N2O reduction occurs at the CuI/CuIV edge (Figure 16A). The μ4SCu4 cluster is held in the protein by 7 His ligands, two on CuI-CuIII and one on CuIV. In the resting crystal structures, there are one or two H2O derived ligands at the CuI/CuIV edge. 36,37
Figure 16.
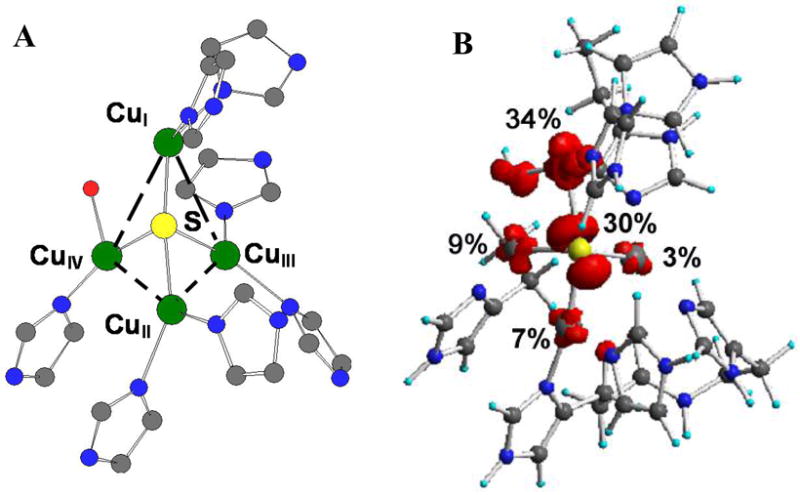
(A) Crystal structure (PnN2OR) (B) Geometry optimized electronic structure
The initial goal of spectroscopy was to determine the electronic structure of the crystallographically defined resting CuZ cluster.38 The CuZ cluster has a sulfide to Cu CT transition at 640 nm which shows a pseudo-A term MCD spectrum. The temperature dependence of the MCD shows that the ground state has Stot=1/2, which can either reflect a site with 1CuII/3CuI or 3CuII/1CuI. These possibilities could be distinguished with XAS at the Cu K-edge, where from Figure 17A the CuZ feature at 8984 eV shows that 3Cus are reduced in resting CuZ.39 This leaves one-hole on the cluster and its distribution could be determined by a combination of X and Q band EPR and S K-edge XAS. Figure 17B shows that the g// value from Q band lies on a Cu hyperfine line in X band requiring e− delocalization over at least 2Cus. These experimental results are supported by geometry optimized DFT calculations (Figure 16B) which show dominant CuI character but significant delocalization of the hole over the cluster depending on the nature of the edge ligand.39
Figure 17.
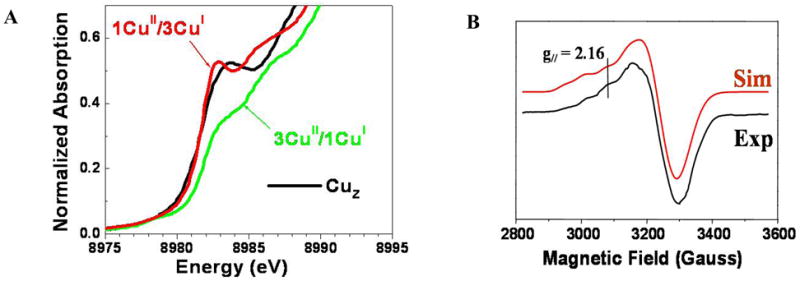
(A) Cu-K edge XAS spectrum of CuZ (black) simulated with 1CuII3/CuI and 3CuII/1CuI (B) EPR spectrum of CuZ from PnN2OR
The edge ligand could be detected by rR spectroscopy which also elucidated the requirement of a proton for high activity of N2OR. A structural model consistent with the results involves an OH− edge ligand whose orientation is affected by H-bonding to a protonated Lys at the CuI/CuIV edge. This protonated Lys appears to play a significant role in assisting catalysis.
We next focus on the redox state of CuZ required for catalysis. Reduction of the CuZ EPR signal of the one-hole form directly correlates with the activity. Thus the fully reduced (4CuI) form of the CuZ reacts with N2O in catalysis.39 Geometry optimization of N2O at the CuI/CuIV edge shows that it binds as a μ-1,3 bridge, bent with an angle of 1390. From Figure 18A this bending greatly lowers the energy of the π* LUMO of N2O which leads to extensive backbonding from fully reduced CuZ into the N2O activating the cleavage of the N-O bond.39
Figure 18.

(A) Backbonding interaction from fully reduced CuZ to N2O (B) N-O bond cleavage barrier of CuZ (red/grey), Cu2SH (black), H-bond assisted (blue).
Figure 18B evaluates the potential energy surface for N-O cleavage on the CuZ cluster. The barrier is reduced from 61 kcal/mol in the gas phase to 18 kcal/mol on CuZ. This is because the bent N2O reactant is destabilized and, in particular, the extensive backbonding lowers the TS energy through stabilization of the CuIVII-O− bond. Allowing for a protonated ligand to H-bond to this oxo further lowers the TS to ~ 10 kcal/mol. Finally since the two e− transferred are donated from CuI and CuIV, it is interesting to evaluate the role of the additional Cus in the cluster. Eliminating CuII and CuIII (and saturating the sulfide by protonation) eliminates much of the backbonding into N2O and increases the barrier for N-O cleavage to 37 kcal/mol.40
The above considerations lead to the molecular mechanism for the 2e−/2H+ cleavage of N2O shown in Figure 19. H-bonding from the protonated Lys lowers the barrier for N-O cleavage which leads to the two-hole hydroxyl bridged species. This is rapidly reduced by CuA to the one-hole species where protonation of the Lys again raises the potential and provides a proton to complete the reaction cycle.40
Figure 19.

N2OR reaction mechanism
Concluding Comments
Spectroscopic/electronic structural studies on copper proteins, their intermediates and models have provided fundamental insights into their reactivities. Among major issues that remain are the structural differences over the coupled binuclear copper proteins that lead to their different reactivities in Figure 1, whether the side-on structure of oxy-Ty directly reacts with substrates or converts to the bis μ-oxo structure along the reaction coordinate of phenolate binding and hydroxylation in Figure 5, the reactivity of the Cu(II) O2- species in H-atom abstraction and its relevance to the PHM mechanism in Figure 11, the fundamental differences of the trinuclear copper cluster in the multi-copper oxidases relative to the coupled binuclear copper protein sites in dioxygen reactivity and the factors involved in the proton assisted reductive cleavage of the O-O bond, and trapping and defining the key intermediates in the reaction mechanism of N2OR in Figure 19. The field of copper bioinorganic chemistry has come a long way, but there is still much to understand.
Acknowledgments
We thank past students and collaborators who have contributed to this work and the NIH (DK31450) for funding.
Works Cited
- 1.Solomon EI, Sundaram UM, Machonkin TE. Multicopper Oxidases and Oxygenases. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 2.Pate JE, Cruse RW, Karlin KD, Solomon EI. Vibrational, electronic. and resonance Raman spectral studies of (Cu2(XYL-O-)O2)+, a copper(II) peroxide model complex of oxyhemocyanin. J Am Chem Soc. 1987;109:2624–30. [Google Scholar]
- 3.Baldwin MJ, Ross PK, Pate JE, Tyeklár Z, Karlin KD, Solomon EI. Spectroscopic and Theoretical Studies of an End-On Peroxide-Bridged Coupled Binuclear Copper(II) Model Complex of Relevance to the Active Sites in Hemocyanin and Tyrosinase. J Am Chem Soc. 1991;113:8671–8679. [Google Scholar]
- 4.Baldwin MJ, Root DE, Pate JE, Fujisawa K, Kitajima N, Solomon EI. Spectroscopic Studies of Side-on Peroxide-Bridged Binuclear Copper(II) Model Complexes of Relevance to Oxyhemocyanin and Oxytyrosinase. J Am Chem Soc. 1992;114:10421–10431. [Google Scholar]
- 5.Metz M, Solomon EI. Dioxygen Binding to Deoxyhemocyanin: Electron Structure and Mechanism of the Spin Forbidden Two-Electron Reduction of O2. J Am Chem Soc. 2001;123:4938. doi: 10.1021/ja004166b. [DOI] [PubMed] [Google Scholar]
- 6.Solomon EI, Baldwin MJ, Lowery MD. Electronic Structures of Active Sites in Copper Proteins: Contributions to Reactivity. Chem Rev. 1992;92:521–542. [Google Scholar]
- 7.Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Oxygen binding, activation, and reduction to water by copper proteins. Angew Chem Intl Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbuhler AD, Tolman WB. Reversible Cleavage and Formation of the Dioxygen O-O Bond Within a Dicopper Complex. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 9.DuBois JL, Mukherjee P, Collier AM, Mayer JM, Solomon EI, Hedman B, Stack TDP, Hodgson KO. Cu K-edge XAS study of the [Cu2(μ-O)2] core: Direct experimental evidence for the presence of Cu(III) J Am Chem Soc. 1997;119:8578–8579. [Google Scholar]
- 10.Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. Spectroscopic and electronic structural studies of the Cu(III)2 bis-μ-oxo core and its relation to the side-on peroxo-bridged dimer. J Am Chem Soc. 1999;121:10332–10345. [Google Scholar]
- 11.Karlin KD, Nasir MS, Cohen BI, Cruse RW, Kaderli S, Zuberbuehler AD. Reversible Dioxygen Binding and Aromatic Hydroxylation in O2-Reactions with Substituted Xylyl Dinuclear Copper(I) Complexes: Syntheses and Low-Temperature Kinetic/Thermodynamic and Spectroscopic Investigations of a Copper Monooxygenase Model System. J Am Chem Soc. 1994;116:1324–1336. [Google Scholar]
- 12.Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. Investigation of the Reactive Oxygen Intermediate in an Arene Hydroxylation Reaction Performed by Xylyl-Bridged Binuclear Copper Complexes. J Am Chem Soc. 1998;120:7841–7847. [Google Scholar]
- 13.Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Tyrosinase Reactivity in a Model Complex: An Alternative Hydroxylation Mechanism. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 14.Klinman JP. Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 15.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: The structure of peptidylglycine alpha-hydroxylating monooxygenase. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 16.Blackburn NJ, Rhames FC, Ralle M, Jaron S. Major changes in copper coordination accompany reduction of peptidylglycine monooxygenase: implications for electron transfer and the catalytic mechanism. J Bio Inor Chem. 2000;5:341–353. doi: 10.1007/pl00010663. [DOI] [PubMed] [Google Scholar]
- 17.Chen P, Bell J, Eipper BA, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine alpha-hydroxylating monooxygenase. Spectroscopic definition of the resting sites and theputative CuM(II)-OOH intermediate. Biochemistry. 2004;43:5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 18.Francisco WA, Wille G, Smith AJ, Merkler DJ, Klinman JP. Investigation of the pathway for inter-copper electron transfer in peptidylglycine alpha-amidating monooxygenase. J Am Chem Soc. 2004;126:13168–9. doi: 10.1021/ja046888z. [DOI] [PubMed] [Google Scholar]
- 19.Chen P, Fujisawa K, Solomon EI. Spectroscopic and theoretical studies of mononuclear copper(II) alkyl- and hydroperoxo complexes: Electronic structure contributions to reactivity. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 20.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. Spectroscopic and electronic structure studies of the diamagnetic side-on Cu-II-superoxo complex Cu(O2)[HB(3-R-5-iPrpz)3]: Antiferromagnetic coupling versus covalent delocalization. J Am Chem Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 21.Chen P, Solomon EI. O2 activation by binuclear Cu sites: Noncoupled versus exchange coupled reaction mechanisms. PNAS. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen binds end-on to mononuclear copper in a precatalytic enzyme complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 23.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. A 1:1 Copper-Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 24.Quintanar L, Gebhard M, Wang TP, Kosman DJ, Solomon EI. Ferrous binding to the multicopper oxidases Saccharomyces cerevisiae Fet3p and human ceruloplasmin: Contributions to ferroxidase activity. J Am Chem Soc. 2004;126:6579–6589. doi: 10.1021/ja049220t. [DOI] [PubMed] [Google Scholar]
- 25.Messerschmidt A, Ladenstein R, Huber R, Bolognesi M, Avigliano L, Petruzzelli R, Rossi A, Finazziagro A. Refined Crystal-Structure of Ascorbate Oxidase at 1.9 Å Resolution. J Mol Bio. 1992;224:179–205. doi: 10.1016/0022-2836(92)90583-6. [DOI] [PubMed] [Google Scholar]
- 26.Quintanar L, Yoon J, Aznar CP, Palmer AE, Andersson KK, Britt RD, Solomon EI. Spectroscopic and electronic structure studies of the trinuclear Cu cluster active site of the multicopper oxidase laccase: Nature of its coordination unsaturation. J Am Chem Soc. 2005;127:13832–13845. doi: 10.1021/ja0421405. [DOI] [PubMed] [Google Scholar]
- 27.Shin W, Sundaram UM, Cole JL, Zhang HH, Hedman B, Hodgson KO, Solomon EI. Chemical and spectroscopic definition of the peroxide-level intermediate in the multicopper oxidases: Relevance to the catalytic mechanism of dioxygen reduction to water. J Am Chem Soc. 1996;118:3202–3215. [Google Scholar]
- 28.Sundaram UM, Zhang HH, Hedman B, Hodgson KO, Solomon EI. Spectroscopic investigation of peroxide binding to the trinuclear copper cluster site in laccase: Correlation with the peroxy-level intermediate and relevance to catalysis. J Am Chem Soc. 1997;119:12525–12540. [Google Scholar]
- 29.Bento I, Martins LO, Lopes GG, Carrondo MA, Lindley PF. Dioxygen reduction by multi-copper oxidases; a structural perspective. Dalt Trans. 2005:3507–3513. doi: 10.1039/b504806k. [DOI] [PubMed] [Google Scholar]
- 30.Rulisek L, Solomon EI, Ryde U. A combined quantum and molecular mechanical study of the O2 reductive cleavage in the catalytic cycle of multicopper oxidases. Inor Chem. 2005;44:5612–5628. doi: 10.1021/ic050092z. [DOI] [PubMed] [Google Scholar]
- 31.Lee SK, George SD, Antholine WE, Hedman B, Hodgson KO, Solomon EI. Nature of the intermediate formed in the reduction of O2 to H2O at the trinuclear copper cluster active site in native laccase. J Am Chem Soc. 2002;124:6180–6193. doi: 10.1021/ja0114052. [DOI] [PubMed] [Google Scholar]
- 32.Aasa R, Branden R, Deinum J, Malmstrom BG, Reinhammar B, Vanngard T. A 17O-effect on EPR spectrum of intermediate in dioxygen-Laccase reaction. BBRC. 1976;70:1204–1209. doi: 10.1016/0006-291x(76)91030-5. [DOI] [PubMed] [Google Scholar]
- 33.Yoon J, Mirica LM, Stack TDP, Solomon EI. Spectroscopic demonstration of a large antisymmetric exchange contribution to the spin-frustrated ground state of a D3 symmetric hydroxy-bridged trinuclear Cu(II) complex: Ground-to-excited state superexchange pathways. J Am Chem Soc. 2004;126:12586–12595. doi: 10.1021/ja046380w. [DOI] [PubMed] [Google Scholar]
- 34.Yoon J, Mirica LM, Stack TDP, Solomon EI. Variable-temperature, variable-field magnetic circular dichroism studies of tris-hydroxy- and μ3-oxo-bridged trinuclear Cu(II) complexes: Evaluation of proposed structures of the native intermediate of the multicopper oxidases. J Am Chem Soc. 2005;127:13680–13693. doi: 10.1021/ja0525152. [DOI] [PubMed] [Google Scholar]
- 35.Cole JL, Ballou DP, Solomon EI. Spectroscopic characterization of the peroxide intermediate in the reduction of dioxygen catalyzed by the multicopper oxidases. J Am Chem Soc. 1991;113:8544–8546. [Google Scholar]
- 36.Zumft WG, Kroneck PMH. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by bacteria and archaea. Advances in microbial physiology. 2006;52:107–227. doi: 10.1016/S0065-2911(06)52003-X. [DOI] [PubMed] [Google Scholar]
- 37.Brown K, Tegoni M, Prudencio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. A novel type of catalytic copper cluster in nitrous oxide reductase. Nat Struct Biol. 2000;7:191–195. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen T, Berks BC, Sanders-Loehr J, Dooley DM, Zumft WG, Thomson AJ. The catalytic center in nitrous oxide reductase, Cuz, is a copper-sulfide cluster. Biochemistry. 2000;39:12753–12756. doi: 10.1021/bi001811i. [DOI] [PubMed] [Google Scholar]
- 39.Chen P, Gorelsky SI, Ghosh S, Solomon EI. N2O reduction by the μ4-sulfide-bridged tetranuclear CuZ cluster active site. Angew Chem Intl Ed. 2004;43:4132–40. doi: 10.1002/anie.200301734. [DOI] [PubMed] [Google Scholar]
- 40.Gorelsky SI, Ghosh S, Solomon EI. Mechanism of N2O Reduction by the μ4-S Tetranuclear Cuz Cluster of Nitrous Oxide Reductase. J Am Chem Soc. 2006;128:278–290. doi: 10.1021/ja055856o. [DOI] [PubMed] [Google Scholar]