

Abstract
Using both rational and random mutagenesis, we have created the first known broad substrate range, nicotinamide cofactor dependent, and highly stereoselective d-amino acid dehydrogenase. This new enzyme is capable of producing d-amino acids via the reductive amination of the corresponding 2-keto acid with ammonia. This biocatalyst was the result of three rounds of mutagenesis and screening performed on the enzyme meso-diaminopimelate d-dehydrogenase. The first round targeted the active site of the wild-type enzyme and produced mutants that were no longer strictly dependent on the native substrate. The second and third rounds produced mutants that had an increased substrate range including straight- and branched-aliphatic amino acids and aromatic amino acids. The very high selectivity towards the d-enantiomer (95 to > 99% e.e) was shown to be preserved even after the addition of the five mutations found in the three rounds of mutagenesis and screening. This new enzyme could complement and improve upon current methods for d-amino acid synthesis.
Introduction
d-Amino acids are increasingly becoming important building blocks in the production of pharmaceuticals and fine chemicals, and as chiral directing auxiliaries, and chiral synthons in organic synthesis. Current important applications of d-amino acids include their use as key components in β-lactam antibiotics, fertility drugs1, anticoagulants2, and pesticides.3 For example, two important d-amino acids used in semi-synthetic antibiotics, d-phenylglycine (ampicillin) and p-hydroxy-d-phenylglycine (amoxicillin), are currently produced on a scale > 5,000 tons per year worldwide.4 In addition, there are more than 20 d-amino acids currently produced at pilot- or full-scale levels, including many d-amino acids that do not have a naturally occurring l-counterpart (such as d-tert-leucine).5 In many of these cases, not only is the d-enantiomer frequently more potent than the corresponding l-enantiomer but it is often more stable in vivo against enzyme degradation, as there are very few metabolic pathways in the liver, kidney and bloodstream capable of catabolizing d-amino acids.
There are currently a number of methods to produce d-amino acids, including enzymatic resolution of the racemate6, enzymatic synthesis using a d-amino acid transaminase7, and the conversion of hydantoins using the coupled enzymatic reactions of a d-hydantoinase and d-carbamoylase.8 While each of these methods are useful, they often have drawbacks including poor yields and enantioselectivity, low reaction rates, low titer of starting material, and the need for multiple enzymatic or chemical reaction steps. The ability to directly aminate a 2-keto acid with ammonia to form a d-amino acid would be highly useful and complementary to the current methods.
As opposed to l-amino acid dehydrogenases, d-amino acid dehydrogenases (d-AADH) are not ubiquitous in nature. In fact, to our knowledge, no broad substrate range, nicotinamide cofactor-dependent, and d-enantioselective amino acid dehydrogenase has ever been reported. d-AADH activity has been observed in a number of bacterial species, including Pseudomonas aeruginosa9, 10, Pseudomonas fluorescens11, Pyrobaculum islandicum12, Salmonella typhimurium13, and Escherichia coli.14-17 However, all these enzymes require dye-based electron acceptors such as methylene blue or 2,6-dichloroindophenol in the oxidative deamination direction. d-Amino acid synthesis via the reductive amination reaction direction was not described. In addition, most of these enzymes are membrane bound, and it would therefore be difficult to obtain high yields of soluble and active enzyme from a suitable expression host such as E. coli. Misono and coworkers have also described the isolation of an NADP+ depended d-theronine deydrogenase from Pseudomonas cruciviae.18 However, this enzyme catalyzes the oxidation of the β-hydroxyl and not the oxidation/reduction of the α-amine and is different from the enzyme described in this work.
In this work we report the creation of the first broad substrate range, nicotinamide cofactor dependent, and highly d-enantioselective amino acid dehydrogenase. This enzyme was created by performing directed evolution on the starting enzyme meso-2,6-d-diaminopimelic acid dehydrogenase (DAPDH) using both rational and random mutagenesis.
Materials and Methods
Materials
NADP+ and NADPH were obtained from BioCatalytics, Inc (Pasadena, CA). d-Amino acids were obtained from Sigma-Aldrich Chemical Co (St. Louis, MO) or Peptech Corp. (Burlington, MA). 2-Keto acids were purchased from Sigma-Aldrich, or if unavailable commercially, were prepared by the addition of the corresponding aldehyde to hydantoin followed by saponification as described previously.19 Nitro Blue Tetrazolium, phenazine methosulfate, and Triton® X-100 were obtained from Sigma-Aldrich. FMOC-Cl (used to derivatize amino acids for UV detection) was purchased from Pierce Biotechnology, Inc. (Rockford, IL). All other chemicals were purchased from Sigma-Aldrich or similar sources and were of reagent grade or higher.
Corynebacterium glutamicum bacteria was purchased from ATCC (Manassas, VA). Platinum pfx DNA polymerase, Fast-Link™ DNA Ligation Kit, pTrcHis2A expression vector and expression host Escherichia coli TOP10 cells were purchased from Invitrogen (Carlsbad, CA). Taq DNA polymerase and Taq DNA polymerase buffer was purchased from Roche (Indianapolis, IN). Restriction endonucleases were obtained from New England Biolabs (Beverly, MA). Oligonucleotides used for PCR amplification were synthesized by IDT Inc. (Coralville, IA). Genomic DNA was isolated with EPICENTRE® (Madison, WI) MasterPure Complete DNA and RNA Purification Kit. Plasmid DNA was purified using PROMEGA (Madison, WI) Plasmid DNA Mini-prep Kit. Gene sequencing was performed by University of Florida, DNA Sequencing Lab (Gainesville, FL). Transformations were performed via electroporation using the MicroPulser (Bio-Rad, Hercules, CA).
Methods
Cloning of the DAPDH gene
The native DAPDH gene (990 bp) was isolated from Corynebacterium glutamicum (ATCC 13032) using standard PCR amplification methods. Based on the published sequence (NCBI accession #Y00151) appropriate primers (forward: 5’-AGTCCCCATGGGTACCAACATCCGCGTAGC-3’, reverse: 5’-AACTGCAGCCCTCGAGGGTTAGACGTCGCGTGCGATCA-3’) were designed incorporating NcoI and XhoI as the 5’ and 3’ cloning sites, respectively. The forward primer also includes a glycine (GGT) after the initial methionine to assist in cloning (all amino acid numberings account for this inclusion). The PCR product was then ligated to an expression vector (pTrcHis2A) cleaved with the same restriction enzymes and transformed into electrocompetent E. coli (TOP10 cell line).
Synthesis of mutant library
Mutant libraries of the DAPDH gene were created using error prone PCR techniques. To accomplish this, 50 pmole of each primer that anneals to the pTrcHis2A vector flanking the DAPDH gene insert (forward:5’-GAGGTATATATTATTGTATCG-3’ and reverse: 5’-GATGATGATGATGGTCGACGG-3’) and 0.1 μg DAPDH/pTrcHis2A plasmid DNA template were added to 1.5 mM MnCl2, 5.5 mM MgCl2, 0.2 mM dATP, 0.2 mM dGTP, 1.0 mM dCTP, 1.0 mM dTTP, 1X Taq polymerase PCR buffer and 2.5 units Taq polymerase. The thermal cycling parameters were 94°C for 2 min (1 cycle), 94°C for 45 s, 52°C for 45 s, 72°C for 90 s (30 cycles), and 72°C for 10 min (1 cycle). The PCR products were digested with DpnI to remove template DNA. The PCR products were purified and digested with NcoI and XhoI. The digested products were then gel-purified and the DAPDH fragments were ligated into the similarly digested vector pTrcHis2A. Ligation mixtures were transformed into E. coli TOP10 cells by electroporation and selected on LB medium supplemented with 100 μg/ml ampicillin.
For site-saturation mutagenesis a similar procedure was used. The PCR reaction included the same forward and reverse primers used to produce the error prone PCR library, internal oligos containing an NNN sequence at the site to be saturated mutagenized with a 15 bp overlap on both sides of the NNN, 0.2 mM dNTP, and 5.5 mM MgCl2. The thermal cycling parameters were identical to those used in the error prone PCR reaction, as was the PCR product purification. The multiple PCR products (0.2 μg/fragment) from this reaction were combined with a second PCR reaction containing 50 pmole of the same forward and reverse primers, 0.2 mM dNTP, and 5.5 mM MgCl2. The thermocycling parameters were: 94°C for 2 min (1 cycle), 94°C for 45 s, 48°C for 45 s, (10 cycles), 94°C for 45 s, 52°C for 45 s, 72°C for 90 s (30 cycles), and 72°C for 10 min (1 cycle). The final PCR product was ligated and transformed as described above.
Screening of mutant library
Individual colonies were picked using an AutoGenesys robotic colony picker (AutoGen, Framingham, MA), into 384-well microtiter plates containing Terrific Broth (TB) media with 100 μg/ml ampilicillin and grown 16 h at 37°C. After growth the master plates were replicated into 384 well plates containing TB, 100 μg/ml ampilicillin, and 50 μM IPTG (to induce gene expression) and allowed to grow 16 h at 30°C. Glycerol was added to the master plates (final concentration of 20%) and stored at −80°C.
After 16 h growth of the replicated plates, the plates were spun down and the supernatant was removed. To the cell pellet, 1 mg/ml lysozyme, 0.1% Triton X-100, 20 mM each of the D-amino acids to be screened, 1 mM NADP+, and 100 mM sodium carbonate, pH 9.5 was added (20 μl/well).
The plates were allowed to shake at 200 rpm. During this time the cells were lysed and the enzyme was allowed to react with the substrate. After 1 h the indicating dye, Nitro Blue Tetrazolium (0.15 mg/ml final concentration) and the electron transfer agent, phenazine methosulfate (0.01 mg/ml final concentration) were added (20 μl/well). The plates were monitored visually for wells changing from pink to purple color. Those wells that turned purple faster than the majority of the wells on the plate (all wells eventually turned purple due to background concentrations of NAD(P)H) were noted as being positives20, 21.
Growth and purification of DAPDH
The wild-type and mutant DAPDH clones were grown and purified using the same procedure. The DAPDH expressing E. coli were grown up in 2.8 L baffled flasks containing 1.2 L TB and 100 μg/ml ampillicin. The flasks were incubated at 30°C and shaken at 180 rpm. After approximately 18 h of growth, the cells were induced with 50 μM IPTG and continued to be incubated at 30°C and shaken at 180 rpm for an additional 18 h. After this time the cells were harvested via centrifugation. The cells were resuspended with 4 ml of 50 mM potassium phosphate buffer, pH 7.5, 0.1 mM DTT, and 0.5 mM PMSF per gram of wet cell paste. Cells were lysed on the APV-1000 homogenizer (Invensys, Albertslund, Denmark) at 13,000 psi. The lysate was treated with 0.15% PEI (50 − 60 KDa molecular weight), 250 mM NaCl, and 50 mM sodium borate to flocculate nucleic acids and lipids, which were then removed by centrifugation. To the treated lysate, solid ammonium sulfate was added to give 45% saturation. After 20 min of stirring, the solids were removed by centrifugation and discarded. The supernatant was brought up to 75% ammonium sulfate saturation and the solids were collected by centrifugation after 20 min of stirring. The pellet was redissolved in 70 ml of 25 mM potassium phosphate buffer, pH 7.5 and ultrafiltrated against 1 L of the same buffer at 4°C. The ultrafiltrated solution was lyophilized and the enzyme was stored at 4°C.
Enzyme assay
The wild-type and mutant DAPDH were assayed spectrophotometrically by monitoring the increase (oxidative deamination reaction direction) or decrease (reductive amination reaction direction) concentration of NADP(H). The typical reductive amination reaction contained the following: 100 mM sodium carbonate buffer, pH 9.0, 200 mM NH4Cl, 25 mM 2-keto acid, and 0.2 mM NADPH. The decrease in absorbance at 340 nm was monitored and an NADPH extinction coefficient of 6.22 mM−1cm−1 was used to correlate absorbance to concentration. The oxidative deamination assay contained 100 mM sodium carbonate buffer, pH 9.5, 25 mM d-amino acid, and 1 mM NADP+. The increase in absorbance at 340 nm was followed and the same extinction coefficient was used. Protein concentration were determined via the Bradford method22 using bovine serum albumin as a protein standard.
Results
First round of mutagenesis and screening: site-directed saturation mutagenesis
For the initial round of directed evolution, we targeted the mutagenesis to those residues that interact with the substituents on the l-carbon in the native substrate, meso-diaminopimelic acid (m-DAP) (see Discussion). Based on structural information determined from X-ray crystallography, these residues have been identified as Arg196, Thr170, and His245.23 To accomplish this, a saturation mutagenesis library was constructed to contain all possible amino acid changes at these three sites. The total number of possible mutants is 203 or 8,000. To increase the likelihood all possible mutations were evaluated a 20,000 colony library was screened. It was also decided to screen substrates that are analogous to the native substrate, mDAP. Table 1 lists the substrates screened against in this library and their similarity to the native substrate. Although the reductive amination reaction direction is the synthetically useful direction, the oxidative deamination reaction was used. This was done since the reduced cofactor product will react (through an electron mediator) with the tetrazolium pro-dye to form a highly colored product. This allows the library to be screened by eye and thus large number of mutants can be screened rapidly. The oxidative deamination enzymatic reaction is reversible, so an enzyme showing activity in this direction will also show activity in the reductive amination direction (with reductive amination being thermodynamically favorable).
Table 1.
Analogs of m-DAP screened against in the saturation mutagenesis library.
| Substrate | Analogy to m-DAP |
|---|---|
 |
Removed amino group on l-carbon |
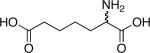
| d,l-2-Aminopimelic acid[a] | |
 |
Removed all charges on l-carbon, kept carbon number the same |
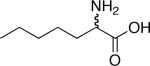
| d,l-2-Aminoheptanoic acid[a] | |
 |
Removed carboxylate group on l-carbon |
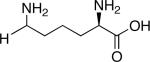
| d-Lysine | |
 |
Removed all charges (amine and carboxylate group) on l-carbon |
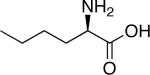
| d-2-Aminohexanoic acid | |
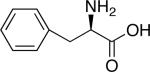 |
Similar in size but with an aryl group |
| d-Phenylalanine | |
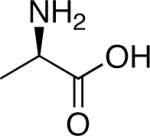 |
Smallest chiral amino acid |
| d-Alanine | |
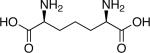 |
Native substrate[b] |
| m-DAP |
These compounds were used in their racemic form due to the unavailability of the pure d-enantiomer.
The native substrate and was not screened against. It is shown here for comparison to the substrates used in the screen.
After screening 20,000 colonies and sub-screening those that were noted as potentially having higher activity, eight were observed to be more active than the wild-type enzyme. After assaying these eight against each substrate separately in the oxidative deamination reaction direction, only d-lysine was found to be an active substrate. Of the eight, one mutant in particular, labeled BC540, had higher activity than the rest. This was sequenced and was found to contain a mutation at each site targeted for mutagenesis: Arg196Met, Thr170Ile, and His245Asn. Interestingly, this mutant no longer exhibited significant activity towards the native substrate, m-DAP.
Second round of mutagenesis and screening
Mutant BC540 was used as the parent for the second round of mutagenesis and screening. In this round random mutagenesis was performed over the entire gene, not just at the active site as was done in the saturation mutagenesis library (first round). This library was screened against the substrates in Table 1 with the exclusion of d-lysine. Since the parent already has activity towards d-lysine, inclusion of it could potentially mask out activity towards any other substrate. After screening approximately 40,000 colonies, 60 were noted as potential positives. These were regrown from the master plate into 12-well plates and assayed against each of the substrates in Table 1 separately, including d-lysine, in the oxidative deamination reaction direction. Four of the 60 showed notably higher activity towards d-lysine, d-norleucine and d,l-2-aminoheptanoic compared to the parent. The most active of these four, mutant BC574 was then assayed towards a number of additional d-amino acids that were not used in the screen in order to ascertain the substrate range of this mutant. Higher activity compared to the parent was observed for d-cyclohexylalanine, d-ornithine, d-2-aminoadipic acid, d-valine, d-leucine, and d-4-chlorophenylalanine. No activity was observed for this mutant or for the parent, BC540, towards d-tert-leucine, d-glutamate, d-aspartate, d-2-naphthylalanine, and d-tyrosine. BC574 was sequenced and was found to have one additional mutation, Gln151Leu, in addition to the three contained in the parent.
Third round of mutagenesis and screening
BC574 was used as the parent for the third and final round of mutagenesis and screening. As in the second round, the mutagenesis was performed randomly over the entire gene. In addition to the substrates listed in Table 1 (excluding d-lysine), this library was also screened against d-cyclohexylalanine, d-ornithine, and d-phenylglycine. After screening 40,000 colonies, 33 potential positives were rescreened after regrowth from the master plate into 12-well plates. Of these 33, seven were measured as having higher activity than the parent for at least one of the substrates screened. These seven were grown up at the 1.2 L scale and purified. At this point these seven mutants were screened against a much broader range of d-amino acids and the activity was measured in the reductive amination direction. This is the synthetically useful reaction direction and is thermodynamically favorable. It has been reported that for the wild-type enzyme and the native substrate (m-DAP) the reductive amination direction is ∼ 100-fold faster than the oxidative deamination direction.24 All seven mutants showed higher activity than the parent for at least one (and in most cases many) substrate(s). However, we found that one mutant in particular, BC621, was the most active and had the broadest substrate range of the seven. BC621 was sequenced and was found to contain one additional mutation compared to the parent, Asp155Gly, giving it a total of five mutations.
Activity of mutant BC621 for d-Amino acid synthesis
The most active DAPDH mutant found after three rounds of mutagenesis and screening, BC621, was assayed for reductive amination activity against a broad range of 2-keto acids for the synthesis of d-amino acids. The wild-type was also assayed under the same conditions for comparison. The results are shown in Table 2.
Table 2.
Activity of mutant BC621 and wild-type DAPDH for the synthesis of D-amino acids by the reductive amination of the corresponding 2-keto acids with ammonia.
| d-Amino acid product | Mutant BC621 (U/mg)[a] | Wild-type (U/mg)[a] | Enhancement over wild-type | Enantiomeric excess |
|---|---|---|---|---|
| d-Alanine (d-2-aminopropionate) | 0.12 | 0.009 | 13 | 77%[e] |
| d-2-Aminobutyrate | 0.25 | 0.018 | 14 | > 99% |
| d-Norvaline (d-2-aminopentanoate) | 0.69 | 0.03 | 23 | > 99% |
| d-Norleucine (d-2-aminohexanoate) | 1.41 | 0.0095 | 257 | > 99% |
| d-2-Aminoheptanoate | 2.15 | 0.004 | 538 | > 99% |
| d-2-Aminooctanoate | 7.8 | 0.008 | 975 | 95% |
| d-Valine | 0.28 | 0.005 | 56 | 98% |
| d-tert-Leucine | N/A[b] | N/A | — | N/A |
| d-Isoleucine | 0.22 | N/A | ∞ | 96% |
| d-Leucine | 0.62 | 0.004 | 155 | > 99% |
| d-Cyclopentylglycine | 0.28 | 0.002 | 140 | > 99% |
| d-Cyclohexylalanine | 2.5 | 0.004 | 625 | > 99% |
| d-Methionine | 1.0 | 0.006 | 153 | > 99% |
| d-Aspartate | N/A | N/A | — | N/A |
| d-Glutamate | 0.025 | N/A | ∞ | N/A |
| d-Phenylglycine | N/A | N/A | — | N/A |
| d-Phenylalanine | 0.11 | N/A | ∞ | > 99% |
| d-Tyrosine[d] | 0.42 | N/D[c] | — | > 99% |
| d-4-Fluorophenylalanine | 0.068 | 0.002 | 34 | > 99% |
| d-4-Chlorophenylalanine | 0.055 | N/A | ∞ | > 99% |
| d-Homophenylalanine | N/A | N/A | — | N/A |
| Control reaction with no 2-keto acid | 0.001 | 0.0004 | — |
1 U = 1 μmol NADP+/min Activity was normalized to unit weight of lyophilized solids.
N/A: No measurable activity was observed with this substrate under the assay conditions used.
N/D: Activity was not determined for this substrate.
Due to the high absorbance of this substrate at 340 nm, activity towards this compound was assayed via HPLC using 25 mM 2-keto acid, 200 mM NH4Cl, 5 mM NADPH, 100 mM sodium carbonate buffer, pH 9.0. The HPLC conditions were the following: Column: Synergi 4μ Polar RP (Phenomenex, Torrance, CA), 250 × 4.6 mm, 4 μ particle size; Program: 1 ml/min of 95% aqueous 20 mM potassium phosphate pH 5.7, 5% MeOH, isocratic; product eluted at 4.5 min and was detected via UV absorbance 225 nm.
Formation of l-alanine due to presence of alanine racemase, see discussion.
Kinetic constants of mutant BC621
The kinetic constants, kcat and KM, for mutant BC621 were measured towards NH4Cl, NADPH and two 2-keto acids. The activity for these experiments was measured spectrophotometrically as described in the Methods section. The high extinction coefficient of NADPH at the λmax of 340 nm precluded us from using this wavelength when measuring the kinetic constants on NADPH. Instead, a wavelength of 375 nm was used; the extinction coefficient at this wavelength was measured to be 1.75 mM−1cm−1. We decided to measure the kinetic constants for the 2-keto acids cyclohexylpyruvate and 2-ketooctanoic acid. The former has been shown to be useful as a pharmaceutical intermediate (component of the thrombin inhibitor inogatran25) while the latter was the substrate mutant BC621 was most active towards. After measuring the initial rates at various substrate concentrations, non-linear regression (using the Microsoft Excel Solver tool) was used to fit the data to the Michaelis-Menten equation. The results of these measurement are shown in Table 3.
Table 3.
Measured kinetic constants of DAPDH mutant BC621[a].
| 2-Keto acid | kcat (U/mg) | KM 2-keto acid (mM) | KM NH4Cl (mM) | KM NADPH (mM) |
|---|---|---|---|---|
| Cyclohexylpyruvate | 9.3 | 41 | 230 | 0.37 |
| 2-Ketooctanoic acid | 11.2 | 15 | Not determined | Not determined |
The substrate concentration range (when determining kinetic constants on this substrate) and static value (when determining the kinetic constants on other substrates) were: 2-Keto acid: 0 − 65 mM, 30 mM NH4Cl: 0 − 1000 mM, 1000 mM NADPH: 0 − 1.0 mM, 0.2 mM
The KM for NH4Cl and NADPH were not determined for 2-ketooctanoic acid but are expected to be similar to those measured for cyclohexylpyruvate. Note that it was difficult to reach saturating conditions for the 2-keto acids and NAPDH due to the high absorbance of these substrates at the concentrations used.
Enantiomeric purity of mutant DAPDH reductive amination product
The enantiomeric selectivity of mutant BC621 was examined. Cyclohexylpyruvate was used as the model substrate and it was aminated with ammonia catalyzed by the mutant BC621. The cofactor, NADPH, was recycled using glucose as the electron source and glucose dehydrogenase as the electron transfer catalysis. The glucose oxidation product, gluconolactone, spontaneously hydrolyzes irreversibly to gluconic acid driving the reaction to completion. This coupled reaction is shown in Scheme 1.
Scheme 1. Reaction scheme for the gram scale synthesis of d-cyclohexylalanine. Reaction conditions.

Buffer: 25 ml of 100 mM sodium carbonate/bicarbonate, pH 9.0
Cyclohexylpyruvate: 1 g (40 g/L, 235 mM)
NH4Cl: 0.63 g (25.2 g/L, 475 mM)
Glucose: 1.6 g (64 g/L, 355 mM)
NADP+: 5.0 mg (0.2 g/L, 0.25 mM)
d-Amino acid dehydrogenase (Mutant BC621): 20 mg (0.8 mg/ml)
Glucose dehydrogenase: 0.25 mg (0.01 mg/ml)
The solution turned cloudy as the reaction proceeded indicating the production of insoluble cyclohexylalanine. After ∼ 6 h, the pH of the solution was lowered to pH 6 causing much of the cyclohexylalanine to precipitate. The resulting cyclohexylalanine was filtered and washed with cold water and dried in a vacuum oven for 24 h. The conversion of substrate was > 95% determined via HPLC, however the isolated yield (unoptimized) was ∼ 70%. The cyclohexylalanine product was dissolved in pH 10 borate buffer and derivitized with FMOC-Cl according to the instructions included with the reagent. The optical purity of the product was determined by chiral HPLC and the resulting chromatograms are shown in Figure 1. The same reaction was performed at 1/25th the scale (1 ml) for each of the 2-keto acids in Table 2 that the mutant DAPDH was active towards (with the exception of 2-keto glutarate, due to the poor activity towards this substrate). After 24 h reaction time the enantioselectivity of the enzyme for each substrate was determined as described previously. These results are shown in Table 2 (see Supporting Information for chiral HPLC conditions).
Figure 1.

Chromatograms of the cyclohexylpyruvate amination reaction product. Top: reaction product; Middle: reaction product spiked with d-cyclohexylalanine standard; Bottom: reaction product spiked with l-cyclohexylalanine standard. The first two peaks in each chromatogram are a result of the FMOC reagent used to derivatize the products for UV detection.
Discussion
The ability to synthesize d-amino acids with high enantiomeric purity via reductive amination of the corresponding 2-keto acid with ammonia is highly desirable due to the low cost of the starting materials. Unfortunately there are no known industrially viable biocatalysts capable of performing this reaction. To solve this problem, we used directed evolution to create a nicotinamide cofactor dependent, broad substrate range, and d-enantioselective amino acid dehydrogenase. A key step in this process is the proper choice of enzyme to perform directed evolution upon. One option is to start with an l-amino acid dehydrogenase and attempt to convert it to a d-amino acid dehydrogenase. While this has been attempted for an hydantoinase26, success was somewhat limited as the enantiomeric excess of the product produced by the mutant enzyme was only 20% for the l-enantiomer (versus 40% for the d-enantiomer of the wild-type enzyme). In the end, we would also expect to find a non-enantioselective enzyme with d-amino acid dehydrogenase activity, rather than a highly enantioselective d-amino acid dehydrogenase.
A better starting choice is the enzyme meso-2,6-diaminopimelic acid d-dehydrogenase. This enzyme catalyzes the reversible oxidative deamination of only the d-amine in meso-2,6-diaminopimelic acid giving l-2-amino-6-oxopimelic acid (Figure 2). It uses NADP(H) as an electron acceptor/donor. Since this enzyme is absolutely selective towards the d-amine in the meso compound27, it can be considered a d-AADH.
Figure 2.

In vivo reaction performed by meso-2,6-diaminopimelate d-dehydrogenase (DAPDH).
DAPDH is found in the lysine biosynthetic pathway where precursors such as l-aspartate and l-homoserine are converted, via multiple steps, into l-2-amino-6-oxopimelate, which is reduced to m-DAP by DAPDH and then enzymatically decarboxylated to l-lysine28. DAPDH has been found in a number of lysine overproducing microorganisms including Bacillus sphaericus29, 30 and Corynebacterium glutamicum31, 32. For our work, we have chosen to use the DAPDH from C. glutamicum. The reasons for this are that the gene sequence32, and crystal structure23 have already been determined, and it has been demonstrated that this enzyme can be cloned into E. coli with high expression and high activity.33 DAPDH is known to be active almost exclusively towards m-DAP; this enzyme exhibits no activity towards a number of alkyl and aryl d-amino acids.29 Therefore, the goal of this work was to broaden the substrate range of this enzyme.
In the first round of mutagenesis and screening, rational mutagenesis was used and specific residues in the active site were targeted. Crystal structure analysis23 has shown that the only interactions to the moieties on the l-carbon are three amino acids that hydrogen bond to the α-carboxylate; no significant hydrogen bonding was found to occur at the α-amine on the same carbon. A library was constructed in which these three amino acids were mutagenized to give all possible amino acid changes. After screening towards analogs of the native substrate, mutants active towards d-lysine were found. Sequencing revealed mutations at all three sites (see Results). The most important mutation is likely Arg195Met. The positive charge on arginine most likely stabilizes the negative charge of the α-carboxyl group on the l-carbon in the native substrate, m-DAP. By removing this charge, the ε-amine on d-lysine would no longer be electrostatically repelled. This also helps explains why m-DAP was no longer a good substrate for the mutant.
Now that the mutant DAPDH was no longer strictly dependent of m-DAP as a substrate, we were successful in finding mutants active towards a broad range of d-amino acids. After the third round of mutagenesis and screening, mutants were found that were able to reductively aminate various aliphatic and aromatic 2-keto acids to produce the corresponding d-amino acids, as described in Table 2. There are a number of interesting observations that can be made from this table. The rate enhancements from the wild-type to the most active mutant can be quite significant – rate enhancements as high as three orders of magnitude were observed. In four cases (d-isoleucine, d-glutamate, d-phenylalanine, and d-4-chlorophenylalanine) mutant BC621 showed activity for a substrate that the wild-type was completely inactive towards. For the unbranched alkyl d-amino acids, the activity of mutant BC621 increased with increasing side-chain length, with 2-aminooctanoate showing the highest activity. The branched and cyclic d-amino acids, with the exception of d-tert-leucine, also showed activity with mutant BC621, albeit not as high as the activity displayed towards some of the unbranched d-amino acids. Poor activity was observed for the acidic d-amino acids assayed. No activity was measurable for D-phenylglycine and the aforementioned d-tert-leucine. This is most likely due to the steric hinderence of the bulky substituents on the α-carbon. However, activity was observed for d-phenylalanine and para-substituted d-phenylalanine. We know now, based on the kinetic information shown in Table 3, that the concentrations of substrates used to generate the data for this table is most likely near or below the KM. Thus, the rates presented therein are likely sub-Vmax rates. For example, the Vmax for d-cyclohexylalanine was determined to be 9.3 U/mg, which is 3.7-fold higher than the activity reported for the conditions used in Table 2.
Enantioselectivity of DAPDH mutant BC621 was determined on all substrates in Table 2 that the enzyme was appreciably active towards. In all cases, except for alanine, the enantiomeric excess of the d-enantiomer > 95% and in most cases it was > 99%. We were unable to accurately measure the e.e. of the alanine reaction due to the apparent presence of an endogenous alanine racemase34. In fact, if we substitute D-alanine for pyruvate in the reaction mixture, we observe the formation of a significant amount of L-alanine, indicating an alanine racemase is present as an impurity in either the mutant DAPDH or the glucose dehydrogenase. In order to use these enzymes for D-alanine synthesis, the alanine racemase would have to be either purified out or selectively inhibited35. The reason for the slight lower e.e. values for some substrates (e.g. 2-aminooctanoate and isoleucine) is not known. However is could be due to an impurity that co-elutes with the L-enantiomer. Control reactions, with no 2-keto acid substrate, also show a peak that elutes at the same time as L-enantiomer. We have been unable so far to resolve the L-amino acid from this impurity. These results demonstrate that even though five mutations were introduced into the wild-type DAPDH, the high selectivity towards the d-enantiomer was preserved.
Conclusions
Through the use of rational and random directed evolution techniques we have created the first known broad substrate range, nicotinamide cofactor dependent, and highly stereo-specific d-amino acid dehydrogenase. This enzyme was found after one round of mutagenesis targeted at the substrate binding site and two rounds of random mutagenesis over the entire gene. Only five mutations were necessary to bring about a significant broadening of the substrate range of the very substrate specific wild-type enzyme. The mutant enzyme exhibits significant activity towards the synthesis of a number of straight, branched and cyclic aliphatic, and aromatic d-amino acids. The mutations did not affect the enantioselectivity of the enzyme as the e.e. value of the d-enantiomer was found to be 95 to > 99% for the mutant enzyme catalyzed reaction product. This enzyme could be very useful in the inexpensive production of highly enantiomerically pure d-amino acids.
Supplementary Material
Footnotes
Supporting Information Available
Chromatography conditions for analyzing the enantiomeric purity of the products produced by the mutant d-amino acid dehydrogenase (BC621). This material is available free of charge via the Internet at http://pubs.acs.org.
Reference Section
- 1.Merviel P, Najas S, Campy H, Floret S, Brasseur F. Minerva Ginecol. 2005;57(1):29–43. [PubMed] [Google Scholar]
- 2.Hauptmann J. Eur J Clin Pharmacol. 2002;57(11):751–8. doi: 10.1007/s00228-001-0392-7. [DOI] [PubMed] [Google Scholar]
- 3.Leahey JP. The Pyrethroid Insecticides. Taylor & Francis; London: 1985. [Google Scholar]
- 4.Schmid A, Hollman F, Park JB, Buhler B. Curr. Opin. Biotech. 2002;13:359–366. doi: 10.1016/s0958-1669(02)00336-1. [DOI] [PubMed] [Google Scholar]
- 5.Bommarius AS, Schwarm M, Drauz K. J. Mol. Catal. B-Enzym. 1998;5:1–11. [Google Scholar]
- 6.Kamphuis J, Meijer EM, Boesten WH, Sonke T, van den Tweel WJ, Schoemaker HE. Ann N Y Acad Sci. 1992;672:510–27. [PubMed] [Google Scholar]
- 7.Galkin A, Kulakova L, Yoshimura T, Soda K, Esaki N. Appl Environ Microbiol. 12. Vol. 63. 1997. pp. 4651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prabhakaran K, George S, Palani P, Ramesh KS, Srinivasan CV, Kandasamy NR, Sadhukhan AK. Process. Biochem. 1993;28(7):447–452. [Google Scholar]
- 9.Bater AJ, Venables WA. Biochim. Biophys. Acta. 1977;468:209–226. doi: 10.1016/0005-2736(77)90115-8. [DOI] [PubMed] [Google Scholar]
- 10.Marshall VP, Sokatch JR. J. Bacteriol. 1968;95:1419–1424. doi: 10.1128/jb.95.4.1419-1424.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukada K. J. Biol. Chem. 1966;241(19):4522–4528. [PubMed] [Google Scholar]
- 12.Satomura T, Kawakami R, Sakuraba H, Ohshima T. J Biol Chem. 2002;277(15):12861–7. doi: 10.1074/jbc.M112272200. [DOI] [PubMed] [Google Scholar]
- 13.Wild J, Walczak W, Krajewska-Grynkiewicz K, Klopotowski T. Mol. Gen. Genet. 1974;128:131–149. doi: 10.1007/BF02654486. [DOI] [PubMed] [Google Scholar]
- 14.Franklin FCH, Venables WA. Mol. Gen. Genet. 1976;149:229–237. doi: 10.1007/BF00332894. [DOI] [PubMed] [Google Scholar]
- 15.Raunio RP, Straus LD, Jenkins WT. J. Bacteriol. 1973;115:560–566. doi: 10.1128/jb.115.2.560-566.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raunio RP, Straus LD, Jenkins WT. J. Bacteriol. 1973;115:567–573. doi: 10.1128/jb.115.2.567-573.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsiewski PJ, Kaczorowski GJ, Walsh C. J. Biol. Chem. 1980;255(10):4487–4497. [PubMed] [Google Scholar]
- 18.Misono H, Kato I, Packdibamrung K, Nagata S, Nagasaki S. Appl Environ Microbiol. 1993;59(9):2963–8. doi: 10.1128/aem.59.9.2963-2968.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Billet G. p-hydroxyphenylpyruvic acid. Organic Syntheses. 1973;Collective volume 5:627. [Google Scholar]
- 20.Mayer KM, Arnold FH. J Biomol Screen. 2002;7(2):135–40. doi: 10.1177/108705710200700206. [DOI] [PubMed] [Google Scholar]
- 21.Williams GJ, Domann S, Nelson A, Berry A. Proc Natl Acad Sci U S A. 2003;100(6):3143–8. doi: 10.1073/pnas.0635924100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradford MM. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 23.Cirilli M, Scapin G, Sutherland A, Vederas JC, Blanchard JS. Protein Sci. 2000;9:2034–2037. doi: 10.1110/ps.9.10.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunhuber NM, Blanchard JS. Crit Rev Biochem Mol Biol. 1994;29(6):415–67. doi: 10.3109/10409239409083486. [DOI] [PubMed] [Google Scholar]
- 25.Gustafsson D, Elg M, Lenfors S, Borjesson I, Teger-Nilsson AC. Blood Coagul Fibrinolysis. 1996;7(1):69–79. doi: 10.1097/00001721-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 26.May O, Nguyen PT, Arnold FH. Nat. Biotechnol. 2000;18:317–320. doi: 10.1038/73773. [DOI] [PubMed] [Google Scholar]
- 27.Misono H, Togawa H, Yamamoto T, Soda K. J Bacteriol. 1979;137(1):22–7. doi: 10.1128/jb.137.1.22-27.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrumpf B, Schwarzer A, Kalinowski J, Puhler A, Eggeling L, Sahm H. J. Bacteriol. 1991;173:4510–4516. doi: 10.1128/jb.173.14.4510-4516.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misono H, Soda K. J. Biol. Chem. 1980;255:10599–10605. [PubMed] [Google Scholar]
- 30.Misono H, Togawa H, Yamamoto T, Soda K. Biochem. Biophys. Res. Comm. 1976;72:89–93. doi: 10.1016/0006-291x(76)90964-5. [DOI] [PubMed] [Google Scholar]
- 31.Wehrmann A, Phillipp B, Sahm H, Eggeling L. J. Bacteriol. 1998;180:3159–3165. doi: 10.1128/jb.180.12.3159-3165.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishino S, Mizukami T, Yamaguchi K, Katsumata R, Araki K. Nucleic Acids Res. 1987;15:3917–3917. doi: 10.1093/nar/15.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy SG, Scapin G, Blanchard JS. Proteins. 1996;25:514–516. doi: 10.1002/prot.12. [DOI] [PubMed] [Google Scholar]
- 34.Walsh CT. J Biol Chem. 1989;264(5):2393–6. [PubMed] [Google Scholar]
- 35.Thornberry NA, Bull HG, Taub D, Wilson KE, Gimenez-Gallego G, Rosegay A, Soderman DD, Patchett AA. J Biol Chem. 1991;266(32):21657–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.