

Abstract
We describe X-ray crystal and NMR solution structures of the protein coded for by Arabidopsis thaliana gene At1g77540.1 (At1g77540). The crystal structure was determined to 1.15 Å with an R factor of 14.9% (Rfree = 17.0%) by multiple-wavelength anomalous diffraction using sodium bromide derivatized crystals. The ensemble of NMR conformers was determined with protein samples labeled with 15N and 13C+15N. The X-ray structure and NMR ensemble were closely similar with r.m.s.d 1.4 Å for residues 8–93. At1g77540 was found to adopt a fold similar to that of GCN5-related N-acetyltransferases. Enzymatic activity assays established that At1g77540 possesses weak acetyltransferase activity against histones H3 and H4. Chemical shift perturbations observed in 15N-HSQC spectra upon the addition of CoA indicated that the cofactor binds and identified its binding site. The molecular details of this interaction were further elucidated by solving the X-ray structure of the At1g77540–CoA complex. This work establishes that the domain family COG2388 represents a novel class of acetyltransferase and provides insight into possible mechanistic roles of the conserved Cys76 and His41 residues of this family.
As part of an ongoing effort to determine the three-dimensional structures of novel eukaryotic proteins, the Center for Eukaryotic Structural Genomics (CESG1) chose the gene product of Arabidopsis thaliana At1g77540.1 for structural characterization on the basis of its target selection algorithm. This protein (At1g77540) consists of 103-residues (11.6 kDa) and had less than 25% sequence identity to any other structure deposited in the Protein Data Bank. In addition, no biochemical function had been established for this protein or its close homologs. However, the Superfamily server (1) predicted a very weak relationship between At1g77540 and proteins of the acyl-CoA N-acyltransferase superfamily (E = 2.5×10−4). GNAT is an extensive superfamily of enzymes that are universally distributed in nature. This diverse set of enzymes uses acyl-CoA to acylate a variety of substrates and has been implicated in a wide range of physiological processes, including DNA transcription, histone acetylation, protein myristoylation, and antibiotic resistance (2). At present, structural information is available in the Protein Data Bank (3) for over forty members of the GNAT superfamily. Typically, these enzymes consist of ~160 amino acids and have a 3-layer (αβα) sandwich architecture consisting of 7 β-strands and 4 α-helices. The fold is conserved despite the rather low pairwise sequence identity (3–23%) of superfamily members (2).
Here we present the X-ray crystal structure at 1.15 Å resolution and the NMR solution structure of the At1g77540. The structures confirm that At1g77540 is a member of GNAT superfamily; specifically, the structures exhibit structural similarity to members of the GCN5-related N-acetyltransferase family of enzymes. We also show that At1g77540 binds acetyl coenzyme A (AcCoA) by mapping interactions of this cofactor with the protein in solution by NMR and by solving the X-ray structure of the At1g77540 protein-CoA complex at 2.06 Å resolution. Finally, we demonstrate that At1g77540 has a detectable level of acetyltransferase activity with histone substrates.
EXPERIMENTAL PROCEDURES
Expression and Protein Purification
Standard CESG pipeline protocols were used for cloning (4), protein expression (5), protein purification (6) and overall information management (7). Briefly, the At1g77540 protein was produced from 2 liters of chemically defined auto-induction medium (8). [U-13C, 15N]-protein for NMR investigations was produced by modifying the auto-induction growth components to include isotopically labeled compounds, 15NH4Cl, [U-13C]-glucose and [U-13C]-glycerol, as the nitrogen and carbon sources. The details of the procedure have been described elsewhere (9).
Protein Crystallization
Crystals of At1g77540 were grown by the hanging drop method from 10 mg mL−1 protein solution in a buffer (50 mM NaCl, 3 mM NaN3, 0.3 mM TCEP, 5 mM MES pH 6.0) mixed with an equal amount of well solution (29% MEPEG 5000, 100 mM sodium citrate, 100 mM PIPES pH 6.5) at 4 °C. Crystals typically grew as square plates with dimensions of about 200 μm × 200 μm × 30 μm. The crystals of At1g77540 belong to space group P21 with unit cell dimensions of a = 27.4 Å, b = 60.6 Å, c = 29.4 Å and β = 91.5°. Crystals were cryoprotected at 4 °C by soaking in a solution containing 30% MEPEG 5000, 100 mM sodium citrate, 100 mM PIPES pH 6.5 supplemented with increasing concentrations of glycerol, up to a final concentration of 20% glycerol. To prepare derivative crystals for phasing purposes, cryoprotected crystals were soaked for additional 40 seconds in the final cryoprotectant solution supplemented with 1 M NaBr.
X-ray Data Collection
X-ray diffraction data for bromide-soaked crystal were collected at sector 32-ID-B of the Argonne National Laboratory Advanced Photon Source (APS). An initial X-ray fluorescence scan of the crystal indicated a maximum of the bromide anomalous signal EBULK-PEAK = 13,479.2 eV and maximum dispersive differences at EBULK-EDGE = 13,477.3 eV. These values, however, characterized the signal of the bulk bromide ions located in the solvent channels of the crystal. It was not immediately clear at what energy the maximal anomalous signal from ordered bromide ions could be obtained. The first dataset was collected at presumed peak energy 13,480 eV. Next, a dataset at high-remote energy of 13,580 eV was collected. Upon completion of data collection an analysis of the amount of anomalous signal in both datasets was performed by using the REFLECTION_STATISTICS utility of PHENIX (10). It became clear that the amount of anomalous signal was higher in the dataset collected at the high-remote energy than in that collected at the presumed peak energy (4.2% vs. 2.8%). Also, a larger number of (in retrospect, correct) anomalous sites were identified by HySS from the high-remote dataset than from the other (5 vs. 3) (11). This observation suggested that the energy maximizing the anomalous signal of ordered bromides was higher than E = 13,480 eV. Therefore, an additional dataset was collected at EPEAK = 13,484 eV. The strategy proved successful: the new dataset had 5% anomalous signal, and 8 (in retrospect, correct) anomalous sites could be identified. Because the crystal showed no detectable radiation damage, additional datasets were collected at the low-remote energy E = 13,380 eV.
X-ray Structure Determination
The datasets of diffraction images were integrated and scaled using HKL2000 (12). The partial bromide substructure of derivatized At1g77540 crystals was determined using HySS (11). The protein structure was phased from 4-wavelength MAD data in CNS (13) using 5 consensus anomalous sites. Initial phase information obtained from CNS to 2.5 Å was further improved and extended to the 1.6 Å resolution of the low-remote dataset by electron density modification. The automatic tracing procedure of ARP/wARP (14) produced an initial model with 86% residues placed of which 93% had side-chains assigned. The structure was completed by alternate cycles of manual building in XFIT (15) and refinement in REFMAC5 (16) against another low-remote dataset extending to 1.15 Å resolution. All refinement steps were monitored by calculating an Rfree value based on 5.1% of the independent reflections. The stereochemical quality of the final model was assessed by PROCHECK (17) and MOLPROBITY (18) (Table 1). Refined coordinates were deposited in RCSB protein databank (3) with accession number 1xmt. The figures were prepared using PYMOL (19).
Table 1.
Crystal Parameters, Data and Refinement Collection Statistics
| Low Remote #2 | Peak | Edge | High Remote | Low Remote #1 | AcCoA complex | |
|---|---|---|---|---|---|---|
| Spacegroup | P21 | P21 | P21 | P21 | P21 | P21 |
| Unit Cell parameters (Å, deg) | a=27.3, | a=27.5, | a=27.5, | a=27.7, | a=27.3, | a=27.9, |
| b=60.6, | b=61.0, | b=61.0, | b=61.5, | b=60.6, | b=63.9, | |
| c=29.4, | c=29.6, | c=29.6, | c=29.8, | c=29.4, | c=29.5, | |
| β=91.5 | β=91.5 | β=91.5 | β=91.5 | β=91.5 | β=90.9 | |
| Data Collection statistics | ||||||
| Energy (keV) | 13.380 | 13.484 | 13.480 | 13.580 | 13.380 | 12.658 |
| Wavelength (Å) | 0.92668 | 0.91953 | 0.91980 | 0.91302 | 0.92668 | 0.97949 |
| Resolution rangea (Å) | 20.29–1.15 (1.18–1.15) | 30.53–1.75 (1.79–1.75) | 30.50–1.60 (1.64–1.60) | 30.73–1.60 (1.64–1.60) | 30.28–1.6 (1.64–1.60) | 31.97–2.05 (2.10–2.05) |
| Reflections (measured/unique) | 113509/32713 | 75129/9963 | 97899/12942 | 98688/13263 | 96220/12701 | 27770/6473 |
| Completeness (%) | 95.9 (70.7) | 99.9 | 99.9 (99.5) | 99.9 (99.4) | 99.9 (99.5) | 99.2 (98.0) |
| Rmergeb | 0.039 (0.345) | 0.049 (0.202) | 0.033 (0.156) | 0.042 (0.212) | 0.027 (0.099) | 0.052 (0.251) |
| Redundancy | 3.5 (1.9) | 7.5 (7.2) | 7.6 (7.3) | 7.4 (6.1) | 7.6 (7.5) | 4.3 (3.9) |
| Mean I/σ (I) | 16.5 (2.2) | 17.8 (6.7) | 26.5 (8.4) | 19.7 (5.8) | 33.6 (13.4) | 18.3 (5.0) |
| Refinement | ||||||
| No. reflections (work/test) | 31030/1658 | 5822/636 | ||||
| Rcrystc | 0.148 (0.290) | 0.160 (0.180) | ||||
| Rfreed | 0.170 (0.318) | 0.220 (0.245) | ||||
| R.m.s.d. bonds (Å) | 0.021 | 0.016 | ||||
| R.m.s.d. angles | 2.022 | 1.664 | ||||
| Average B factor (Å2) | 17.8 | 34.0 | ||||
| No. water molecules | 125 | 61 | ||||
| No. ions/cofactors | 11 bromides | 1 CoA | ||||
| PDB code | 1XMT | 2GDB | ||||
| Ramachandran plot, residues in | ||||||
| Most favorable region (%) | 91.6 | 89.9 | ||||
| Additional allowed region (%) | 8.4 | 10.1 | ||||
| Generously allowed region (%) | 0.0 | 0.0 | ||||
| Disallowed region (%) | 0.0 | 0.0 | ||||
Values in parentheses are for the highest resolution shell.
Rmerge = ΣhΣI|Ii(h) - <I(h)>|/ΣhΣiIi(h), where Ii(h) is the intensity of an individual measurement of the reflection and <I(h)> is the mean intensity of the reflection.
Rcryst = Σh| |Fobs|−|Fcalc| |/Σh|Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively.
Rfree was calculated as Rcryst using 5.0% of the randomly selected unique reflections that were omitted from the structure refinement.
Structure of At1g77540–CoA Complex
At1g77540 crystals were soaked overnight at 4 °C in 29% MEPEG 5K, 100 mM sodium citrate, 100 mM PIPES pH 6.5 supplemented with 9 mM acetyl-CoA. The crystals were cryoprotected with Fomblin 2500 (Aldrich, Saint Louis, MO). X-ray diffraction data for the complex were collected at NE-CAT 8BM beamline at APS and processed with HKL2000. The structure of the complex was solved by molecular replacement in MOLREP (20), manually adjusted with COOT (21) and refined in REFMAC5.
NMR Data Collection
NMR spectra used for the structure determination were collected at 25 °C on Varian INOVA 600 MHz instruments equipped with a 5 mm z-shielded gradient 1H-13C-15N triple resonance probe at NMRFAM (University of Wisconsin, Madison). The NMR sample conditions for At1g77540 consisted of ≈1 mM protein in 10 mM KH2PO4, 50 mM KCl, 3 mM NaN3, 10% D2O, pH 6. NMR data sets used for backbone resonance assignments included: 2D 1H-15N HSQC, 3D HNCO, 3D HNCACB, 3D CBCA(CO)NH. NMR data sets used for side chain assignments included: 2D 1H-13C HSQC, 3D HBHA(CO)NH, 3D HC(CO)NH, 3D C(CO)NH, 3D H(C)CH TOCSY and 3D (H)CCH TOCSY. All NMR spectra were processed with NMRPipe (22) and with NMRView (23). Spectra used in the CoA binding study were recorded at 25 °C on a 500 MHz Bruker DMX spectrometer equipped with a triple-resonance CryoProbe™ at NMRFAM. The spectra were processed and analyzed using the Bruker XINNMR software.
NMR Structure Determination
The initial 1H-15N HSQC spectrum revealed resonances of uniform shape and intensity consistent with a well folded protein. The At1g77540 protein studied consisted of 103 residues with 6 internal prolines. Of 97 backbone amide cross peaks expected in the 1H-15N-HSQC spectrum, 93 were observed; thus the protein was thus considered to be amenable to high-throughput NMR structure determination. Two residues, Gly50 and Gly52 displayed extremely weak cross-peak intensity in the 1H-15N-HSQC spectrum. The automated assignment program PISTACHIO (24) was used to assign 95% of backbone resonances and 70% of 13C-side chain resonances. The remaining resonances were assigned by standard manual NMR methods. Secondary structural elements were identified by combined analysis of assigned chemical shifts and patterns of NOEs from the 15N- and 13C-edited NOESY spectra.
NMR Structure Calculation
Initial NOE assignments in the 15N-edited NOESY-HSQC (τmix = 100 ms) and 3D 13C-edited-NOESY-HSQC (τmix = 100 ms) data sets, including those indicative of secondary structural elements, were performed manually. These assignments were then used as preliminary input for automated NOE assignments. Structure calculations were performed with CNS (13) using ARIA 1.2 setup and protocols (25, 26). Subsequent addition and/or correction of NOE assignments were performed manually. One-hundred conformers were generated initially, and the best 20 of these, selected by the ARIA algorithm on the basis their agreement with distance input restraints, were used for NOE calibration and assignment. This iterative process was repeated until a final calculation produced 20 converged, low energy structures. A total of 1925 unambiguous nuclear Overhauser effect derived restraints (NOEs) and 172 ambiguous NOEs, assigned by ARIA1.2 or by manual intervention, served as the foundation of the final structure calculation (Table 2). Additional terms in the calculation included 87 dihedral angle constraints derived from TALOS (27) and 56 hydrogen bond constraints derived from a combination of CSI and NOE analysis. These twenty structures were subjected to a final refinement protocol in which a physical force field and explicit water solvent were added to experimental restraints within the ARIA program. The final twenty structures of At1g77540 were validated by PROCHECK NMR (17). The NMR structure was deposited in the Protein Data Bank (3) under PDB # 2EVN; NMR data were deposited in the BioMagResBank (28) under BMRB # 6338.
Table 2.
Statistics for the 20 Conformers Representing the NMR Solution Structure of At1g77540
| Root-mean-square-deviations relative to average structure (Å) | |
| Backbone (Cα, C′, N) atoms in 2nd structure | 0.23 ± 0.04 |
| Backbone all residues | 1.18 ± 0.51 |
| Heavy atoms in 2nd structure | 0.57 ± 0.08 |
| Heavy atoms in all residues | 1.47 ± 0.41 |
| Number of experimental restraints | |
| Intra residue NOEs | 823 |
| Inter residue sequential NOEs ( |i−j| = 1) | 486 |
| Inter residue medium range NOEs ( 1 < |i−j| < 5) | 333 |
| Inter residue long range NOEs ( |i−j| > 4) | 455 |
| Total NOEs | 2097 |
| Dihedral angle restraints | 87 |
| H-bond restraints | 52 |
| Restraint violations | |
| NOE distances with violations > 0.3Å | 0.45 ± 0.67 |
| dihederal with violations > 3° | 1.57 ± 0.74 |
| H-bond with violations > 0.3Å | 0.0 ± 0.0 |
| Final energies from simulated annealing (kcal/mol) | |
| Fvdw | −931 ± 25 |
| Fele | −3785 ± 70 |
| Deviation from idealized geometry | |
| Bonds (Å) | 0.0045 ± 0.001 |
| Angles (deg) | 0.60 ± 0.02 |
| Impropers (deg) | 1.90 ± 0.06 |
| Ramachandran analysis (% of all residues) | |
| Residues in most favored regions | 76 |
| Residues in additional allowed regions | 17.9 |
| Residues in generously allowed regions | 3.52 |
| Residues in disallowed regions | 2.57 |
CoA NMR Titration
A 1H-15N-HSQC spectrum of At1g77540 collected under the above solution conditions was used for reference. A 50 μl aliquot of a 6 mM stock solution of CoA was added to the protein solution to make the 1:1 (CoA:At1g77540) mixture. The pH was adjusted to 6.0 by addition of 3 μl of 0.1 M HCl. A 1H-15N-HSQC spectrum of this sample was then collected under conditions identical to the reference.
Acetyltransferase Assays
Aliquots of At1g77540 were dialyzed for 6 h into buffer containing 50 mM Tris pH 7.0, 150 mM NaCl, and 1 mM DTT. The protein concentration after dialysis was determined by Bradford assay to be 4.4 mg mL−1. Aliquots were frozen in liquid nitrogen and stored at −20 °C until use. Acetyltransferase activity was measured by using a filter-binding assay as described previously (29). Briefly, reactions were carried out in 50 mM Tris, 50 mM Bis-Tris, 100 mM acetate buffer (1x TBA) pH 7.0 containing 1 mM DTT, 100 μM acetyl-CoA (4.95 μCi [3H]-acetyl-CoA), and 6 μM–5 mM amino acid or peptide substrate. Enzyme concentration in the final reaction mixtures was 0.08 mg mL−1. Assays were performed at 25 °C. Data were visualized in KaleidaGraph (Synergy Software, Reading, PA).
RESULTS
At1g77540 Structure Statistics
The crystal structure of At1g77540 was solved to a resolution of 1.15 Å by four-wavelength multiwavelength anomalous diffraction (MAD) using sodium bromide derivatized crystals. Data collection, refinement and model statistics are summarized in Table 1. The final model describes a monomer in an asymmetric unit, containing residues 5–99. In addition, 11 bromide ions and 125 water molecules were built into the final structure.
Independently and in parallel to the crystal structure, we solved the solution NMR structure of At1g77540. A summary of experimental restraints and statistics for the final ensemble of conformers is shown in Table 2. The root-mean-square deviation (r.m.s.d.) for backbone atoms of residues 8–98 of the final ensemble relative to the average structure was 0.36 Å. Measurements of amide backbone 15N-T2 relaxation times (data not shown) gave an average value of 88 ± 15 ms. This value is consistent with At1g77540 being monomeric in solution; this fact is also corroborated an apparent monomeric state of At1g77540 in the crystals.
At1g77540 Fold
The three-dimensional structure of At1g77540 revealed that this protein belongs to the αβ-class of proteins with 2-layer (αβ) sandwich architecture (Figure 1A). The central feature of this protein is a mixed 5-stranded β-sheet formed by four antiparallel β-strands (A, B, C and D), and β-strand E parallel to β-strand D (Figure 1A). A single turn 310-helix 1 forms in the loop connecting β-strands A and B. A short α-helix 2 is located in the loop connecting β-strand D and the central “signature” α-helix 3, which is grasped by the convex surface of the central β-sheet. Two additional helices 4 and 5 are located in the carboxyl-terminal part of the protein. Helix 4 is aligned at approximately 90° relative to helix 3.
Figure 1.
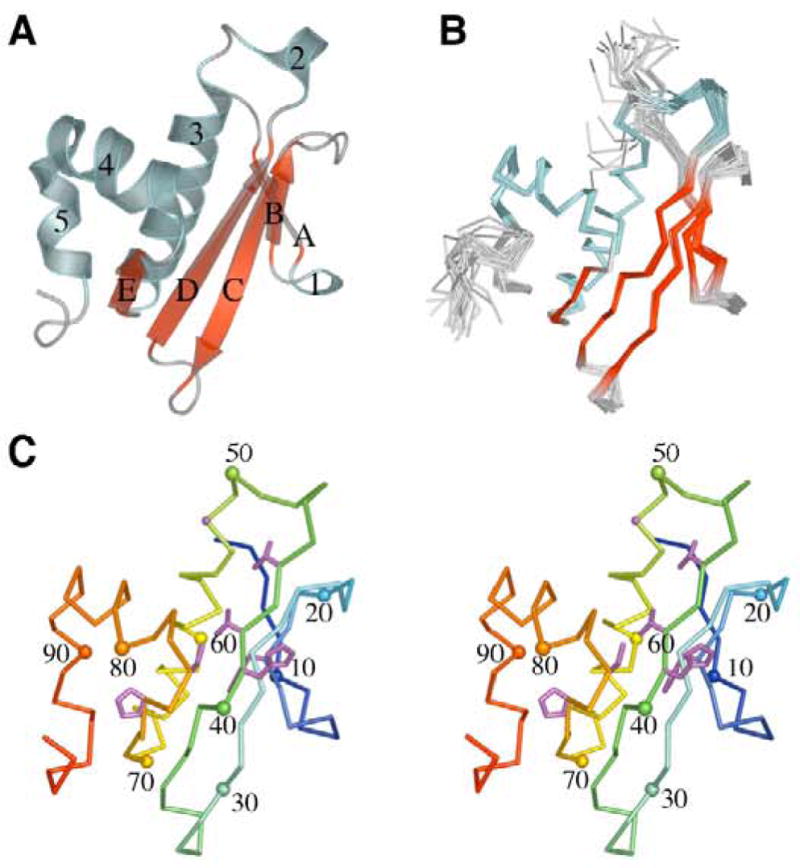
Structure of At1g77540 from A. thaliana. (A) Ribbon diagram of the crystal structure of At1g77540 showing its 2-layer (αβ) sandwich architecture consisting of 5 β-strands (A–E), four α-helices (2–5), and one 310-helix (1). (B) Overlay of backbone traces from the top 20 conformers representing the NMR solution structure of At1g77540. (C) Stereoscopic view of the backbone of At1g77540 color coded from amino-terminus (blue) to carboxyl-terminus (red). Every tenth Cα is labeled and highlighted by a sphere of a color corresponding to that of the backbone. Highly conserved residues (shown as ball-and-stick, blue) include Phe17, His41, Thr42, Val44, Gly50, Gly52, Cys76, and Pro74.
Comparison of X-ray and NMR Structures
A ribbon trace of the final ensemble of 20 NMR conformers chosen to represent the solution structure is shown in the same orientation as the crystal structure of At1g77540 (Figure 1B). Comparison of the X-ray structure and the representative NMR structure over all backbone heavy atoms in the structured residues 8–93 gave an r.m.s.d. of 1.4 Å. This indicates that the overall folds are in good agreement. Slight differences in the two structures were observed in the regions of defined secondary structure. Whereas all elements of secondary structure calculated in the NMR study were observed in the X-ray structure, the two single-turn helices 1 and 2, and helix 5 seen in the X-ray structure (Figure 1B) were not defined in the NMR structure (Figure 1). These regions span residues 12–14 in the loop between β-strands A and B, residues 45–48 following the β-strands D, and residues 88–93 close to the carboxyl-terminus of the protein. (For better orientation a stereo ribbon trace of the X-ray At1g77540 structure with every 10th residue highlighted is presented in Figure 1C.) The lack of clear secondary structure in these regions of the NMR structure likely stems from backbone flexibility, as often is observed in loops and termini of proteins in solution. Recalculation of the backbone r.m.s.d. between the X-ray and NMR structures using only those residues in conserved secondary structural elements revealed a value of 1.2 Å, which supports the notion that the deviation in the backbone coordinates stems from inherent flexibility in the loops and the carboxyl-terminal region of the protein. Several amino- and carboxyl-terminal residues (1–5 and 99–103, respectively) are poorly defined in the NMR ensembles. These segments correlate well with highly disordered regions that lacked interpretable electron density in the At1g77540 X-ray map.
Sequence Homology
Analysis of the At1g77540 sequence by profile alignment methods revealed that this protein shows very weak similarity to the acyl-CoA N-acyltransferase superfamily (E = 5.4×10−5) (1). A Conserved Domain Database (30) search suggested that At1g77540 belongs to the COG2388 family of predicted acetyltransferases (E = 6×10−4). The COG2388 family is related to Pfam00583, which is known as the GCN5-like N-acetyltransferase (GNAT) family. The GNAT family members are important for the regulation of cell growth and development; many of them are involved in the histone acetylation and chromatin remodeling (2). Sequence alignment of twenty COG2388 family members revealed two absolutely conserved residues, Gly52 and Cys76, and several highly conserved residues, including Phe17, His41, Thr42, Val44, Gly50, and Pro74 (Figure 2). All these residues are shown in Figure 1C in ball-and-stick representation. Residue Gly52, which corresponds to the key residue conserved in the GNAT superfamily, is located in the conserved (Arg/Gln)-X-X-Gly-X-(Gly/Ala) motif, which is implicated in the binding of acetyl-CoA (31). The motif is located in the loop connecting helices 2 and 3 (Figure 1A) of At1g77540 that contains residues Arg49-Gly50-Leu51-Gly52-Leu53-Ala54. The importance of Cys76 and His41 is discussed below.
Figure 2.

Sequence alignment of the COG2388 family. Multiple sequence alignment of At1g77540, a putative N-acetyltransferase from Staphylococcus aureus (PDB # 1r57), the histone acetyltransferase Hat1 from Saccharomyces cerevisiae, and 20 seed members of the COG2388 family from the Conserved Domain Database (30). The GenBank accession codes for these seed members are listed in the left column. Secondary structural elements of At1g77540 are represented by cylinders (helices) and arrows (β-strands). The ruler indicates every 5th (*) and 10th (|) residue of At1g77540 is provided for orientation. The number of proteins residues of selected proteins not included in the alignment is shown in brackets. Boxed and shaded areas correspond to motif A (yellow), motif B (pink), and motif D (green) defined for the GNAT family proteins (35) from the structural alignment of At1g77540 and Hat1 (PDB # 1bob). Fully and highly conserved residues are highlighted by bold red letters.
Structural Homology
To confirm that At1g77540 belongs to the GNAT superfamily we submitted the 3D structure to the DALI (32) and VAST servers (33). The top homologs identified by both servers were, indeed, established or hypothetical members of the GNAT superfamily. The closest structural homolog found by DALI was a hypothetical Gcn5-like N-acetyltransferase from Staphylococcus aureus with Z-score of 10.7, r.m.s.d. 2.6 Å and 25% sequence identity over 87 aligned residues (PDB # 1r57). The top homolog identified by VAST was a histone acetyltransferase Hat1 from Saccharomyces cerevisiae with VAST-score of 9.3, r.m.s.d. 1.6 Å and 12.5% identity over 72 aligned residues (PDB # 1bob; (34)). At1g77540 showed significant structural similarity to more than forty additional established or putative acyltransferases. Interestingly, not a single residue within the structurally aligned regions of At1g77540 and its top ten homologs identified by VAST was fully conserved. This suggests that very diverse protein sequences can result in the fold adopted by the GNAT superfamily.
Structural Alignment
A structural alignment of At1g77540 and Hat1, the top homolog identified by VAST, revealed that both structures contain the “signature” helix wrapped by a central β-sheet (Figure 3a) characteristic of the GNAT superfamily (2). Also, both proteins contain a short carboxyl-terminal β-strand (E in At1g77540) juxtaposed in antiparallel direction to a β-strand (D in At1g77540), which possesses the universally conserved β-bulge (2). The backbone amide of the residue next to the β-bulge (analogous to Thr42 in At1g77540) was found to form a hydrogen bond to the carbonyl oxygen of the thioester group of bound acetyl-CoA. At1g77540 and Hat1 differ substantially in the amino- and carboxyl-terminal regions. The amino-terminal region of Hat1 (residues 1–121, not shown in Figure 3a) forms an independent domain attached to the central acetyltransferase domain by a β-strand antiparallel to the β-strand that corresponds to A in At1g77540. In comparison to At1g77540, Hat1 has a much longer carboxyl-terminal region, which forms a helical-bundle-like satellite domain (Figure 3a). In addition, structural elements corresponding to motif C, a conserved sequence previously defined for GNATs (35, 36), are missing in At1g77540. In Hat1 and other GNATs this motif forms a long helix that runs perpendicular to the strands of the central β-sheet. In At1g77540, this is represented only by a short connection between residues 13 and 15 (Figure 3a). The absence of structural elements corresponding to motifC and the additional upstream residues in At1g77540 leaves one face of the central β-sheet fully exposed (Figure 3a). The remaining sequence motifs A, B, and D defined for GNATs (35, 36) are structurally present in At1g77540 (Figure 2). It thus appears that At1g77540 presents a “minimal” version of the acetyltransferase domain. In summary, fold analysis by DALI and VAST as well as the detailed inspection of structural features constitutive for acetyltransferases confirmed that At1g77540 is a member of the GNAT superfamily (2).
Figure 3.
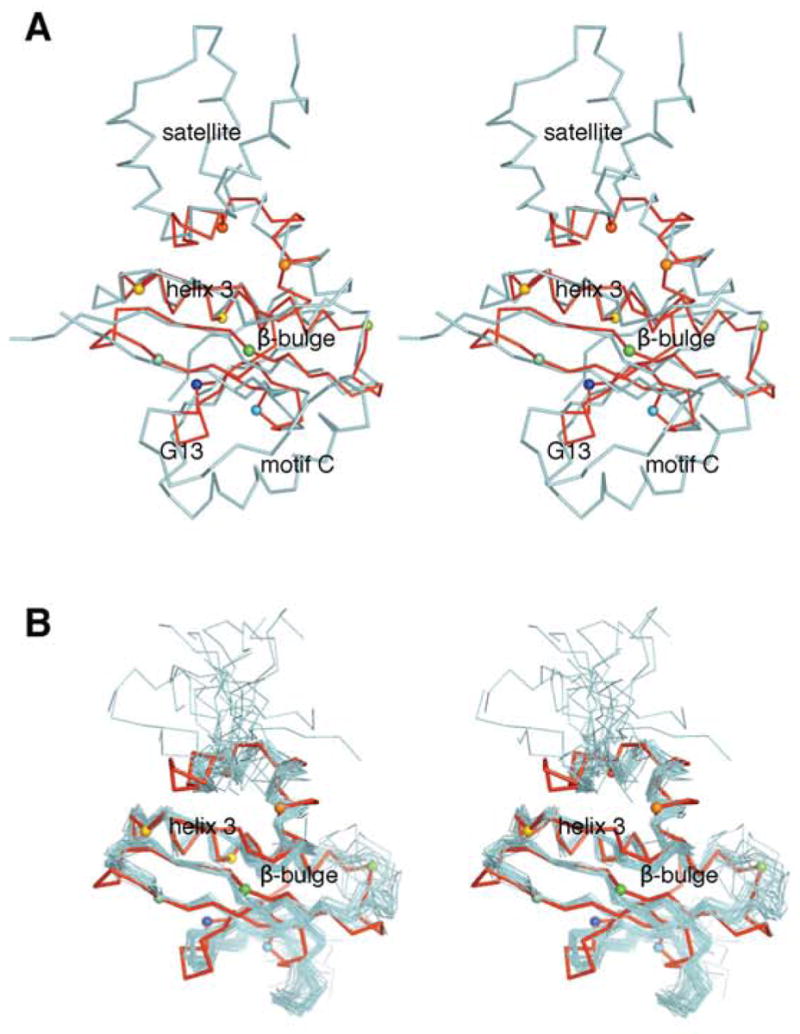
Structural alignment of At1g77540 with (A) Hat1. Stereo diagram showing the aligned structural models for At1g77540 (red; PDB # 1xmt, this study) and histone acetyltransferase Hat1 from Saccharomyces cerevisiae (cyan; residues 122–300; PDB # 1bob). (B) COG2388 family member 1r57. Stereoscopic diagram showing the aligned structural models for At1g77540 (red; PDB #1xmt) and closest structural COG2388 family member from Staphylococcus aureus (PDB # 1r57). The view is rotated approximately 90° clockwise compared to that in Figure 1. Selected Cα atoms are highlighted by spheres, with colors consistent with those used in Figure 1C. At1g77540 contain only a minimal acetyltransferase fold with the conserved “signature” helix 3, central β-sheet containing functionally important β-bulge, and the CoA-binding loop (residues 43–53). Important structural features present in Hat1 but not present in At1g77540 include 1) motif C, which forms a helix that runs perpendicular to the β-strands of the central β-sheet; 2) the independent amino-terminal domain of Hat1 (residues 1–121 of Hat 1, not shown); 3) the helix-bundle-like satellite domain at the carboxyl-terminus of Hat1.
Structural alignment of At1g77540 with the only other COG2388 family member (PDB # 1r57) is displayed in Figure 3b. The two proteins share an identical fold and both reveal the common β-bulge, a structural trait which is common for GNAT superfamily members.
Acetyltransferase Activity Measurements
Both the sequence and structure homology searches suggested that At1g77540 is likely an acetyltransferase with unknown substrate specificity. To test whether At1g77540 functions as a protein acetyltransferase, we performed enzymatic assays utilizing radiolabeled acetyl-CoA and various substrates. The results showed that At1g77540 displays substrate specificity, because some substrates were acetylated while others were not. Observed acetylation of histones H3 and H4 as substrates (Figure 4) suggests that At1g77540 catalyzes protein acetylation. The rate determined for histone H3 was 0.04 nmol min−1 mg−1 enzyme and for histone H4 was 0.015 nmol min−1 mg−1 enzyme. At1g77540 did not exhibit significant acetyltransferase activity with a number of other substrates tested (data not shown): tubulin, lysine, BSA, histones H2A, and H2B, tubulin, and other small histone-based peptides.
Figure 4.
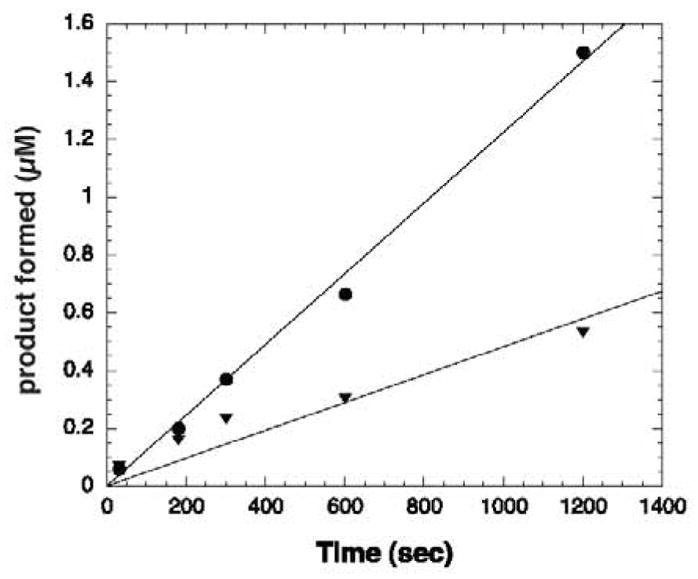
Acetyltransferase activity assay of At1g77540. At1g77540 displayed acetyltransferase activity on histones H3 and H4. Data were generated using a filter binding assay (29). 0.08 mg/mL enzyme was mixed with 1 mM DTT, 100 μM acetyl-CoA (4.95 μCi [3H]-acetyl-CoA), and 100 μM peptide substrate in 1× TBA buffer pH 7.0. Data were graphed as product formed vs. time, and a linear fit was applied to determine the rate. At1g77540 acetylates H3 (circles) at a rate of 0.04 nmol min−1 mg−1 of enzyme and H4 (triangles) at a rate of 0.015 nmol min−1 mg−1 of enzyme. The rate of non-enzymatic acetylation was determined from a matching reaction that contained enzyme buffer instead of enzyme. The background rate was determined and subtracted from the enzymatic rate. KaleidaGraph was used to analyze and plot the data.
Mapping of the CoA-binding Site
To provide evidence that At1g77540 binds (acetyl)-CoA in solution and to map the (acetyl)-CoA binding site, we compared chemical shifts from 1H-15N-HSQC spectra collected in the absence and presence of an equimolar amount of CoA. This strategy can be rationalized because motif A of the GNAT family, which is structurally conserved in At1g77540, supports the binding of acetyl-CoA through a network of hydrogen bonding interactions to backbone amide groups (37). Therefore, interactions between At1g77540 and CoA should be manifested by changes in resonance positions of backbone amide groups. The assigned HSQC spectrum of At1g77540 is shown in Figure 5A. Overlay of 1H-15N-HSQC spectra collected in the absence and presence of equimolar CoA revealed that the positions of 24 NH-backbone resonances are affected by CoA addition (Figure 5B). The affected residues were mapped onto the surface of At1g77540 (Figure 6A). The vast majority of these residues are located within the cleft formed by the carboxyl-terminal end of β-strand D (reside 42 and 44), the following loop (residues 47, 50–52), N-terminal region of helix 3 (residues 54, 55, 57 and 58), and helix 4 (residues 76, 78, 82–84, and 86).
Figure 5.
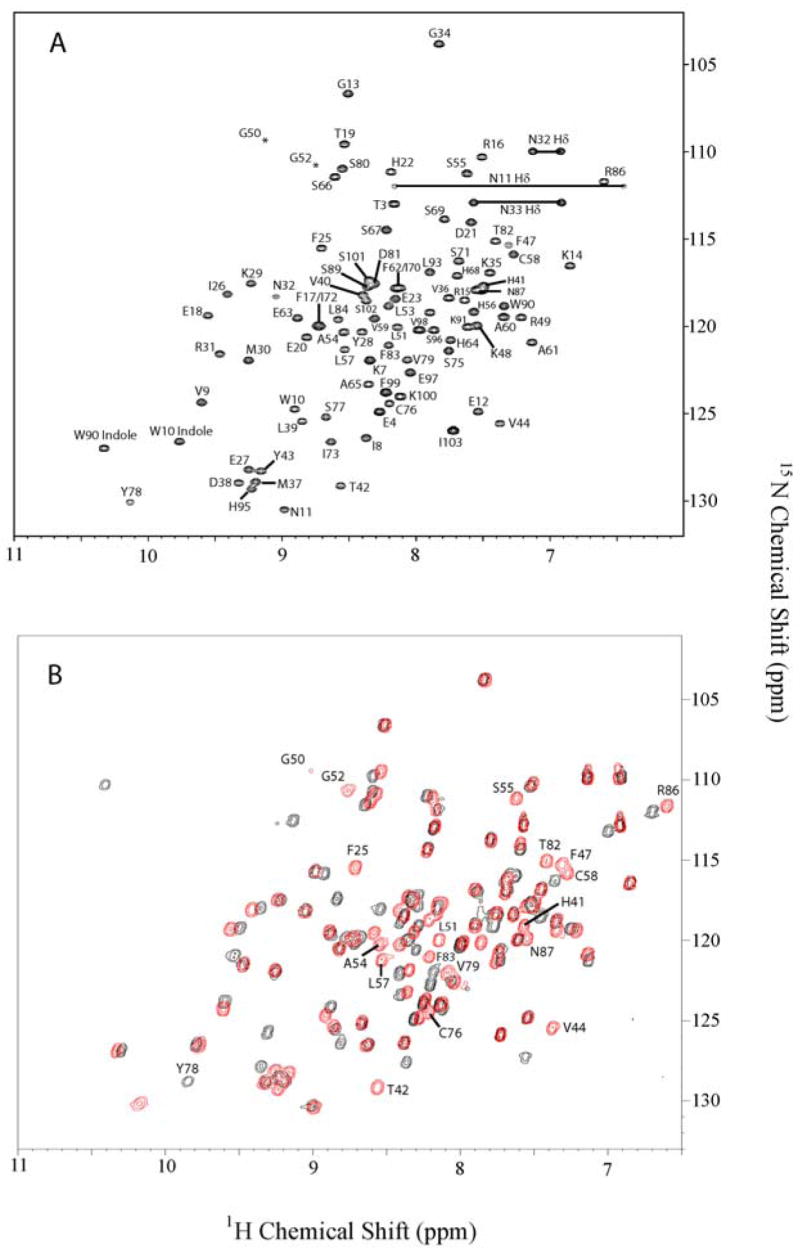
1H-15N-HSQC spectrum of free and CoA-bound At1g77540. A, Spectrum of uniformly 15N-labeled At1g77540 recorded at 25°C in 10 mM KH2PO4, 50 mM KCl, 3 mM NaN3, and 90% H2O/10% D2O at pH 6.0. Assigned backbone amide resonance peaks are labeled with the single letter amino acid abbreviation and residue number corresponding to the primary sequence. Side chain indole resonances for Trp10 and Trp90 are shown in the lower left hand corner of the spectrum. Horizontal lines in the upper right hand corner display positions of side-chain amide groups of residues (Asn11, Asn32, and Asn33). B, Overlay of 1H-15N-HSQC spectra At1g77540 (red) and At1g77540–CoA complex (black). The most prominent chemical shift changes of residues affected by the addition of equimolar CoA to the protein solution are labeled as single letter abbreviation.
Figure 6.
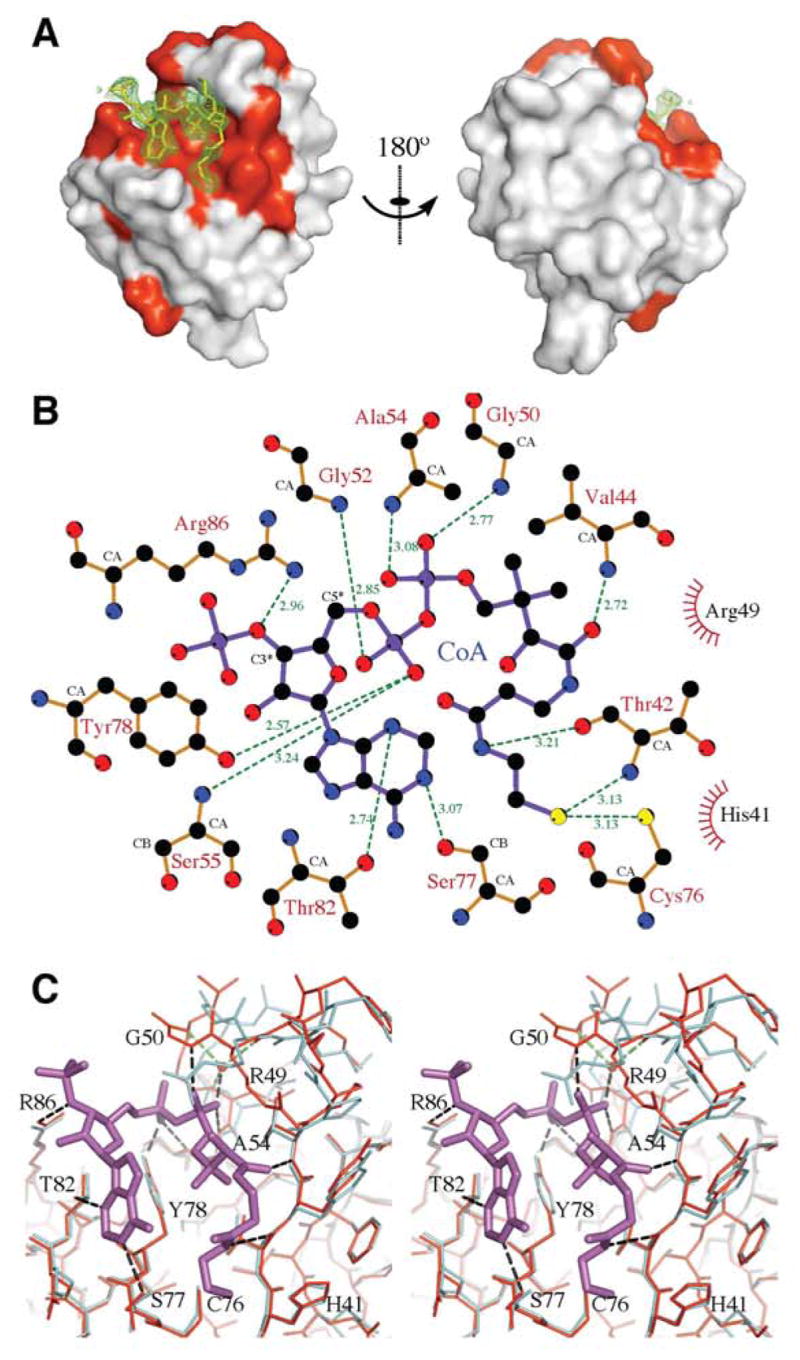
Structural details of CoA binding to At1g77540. (A) The 25 residues significantly affected by the addition of CoA to At1g77540 in solution, as judged from spectral changes in 1H-15N-HSQC spectra, are highlighted in red and mapped onto the surface of the X-ray derived model for the At1g77540-CoA complex. CoA in the complex is indicated by yellow sticks. The omit map for CoA, contoured at a level of 0.6 σ, is shown by green mesh. (B) Schematic diagram of At1g77540 interactions with CoA. Hydrogen bonds are represented by dashed lines; interatomic distances are given in angstroms. “Radiating” spheres indicate hydrophobic contacts between the cofactor and the surrounding residues. The scheme was generated by LIGPLOT (45). (C) Overlay of the X-ray structural models of At1g77540 (cyan) and At1g77540-CoA complex (red). CoA is shown in magenta. Hydrogen bonds are represented by black dashed lines. Selected residues are labeled for orientation.
At1g77540-CoA Complex
We subsequently characterized the complex by reference to the 2.06 Å resolution X-ray structure of At1g77540 crystals soaked in acetyl-CoA. Data collection, refinement and model statistics are summarized in Table 1. As expected, the cofactor bound within the cleft identified by the NMR experiment described above (Figure 6A). The bound cofactor adopts a U-like overall conformation that is stabilized by several hydrogen bonds to At1g77540, electrostatic interactions with charged residues, and hydrophobic contacts (Figure 6B). Specifically, the side chains of Ser77 and Thr82 form hydrogen bonds to the adenine ring; the side chain of Arg86 is in electrostatic contact with the 3′-phosphate group; the side chain of Tyr78 and the amide nitrogen of Gly52 and Ser55 form hydrogen bonds to the 5′-phosphate group; the amide nitrogens of Gly50 and Ala54 and a water molecule, which can form hydrogen bonds to the polar backbone atoms of any of residues Lys48, Leu51, Gly52, and Leu53, form hydrogen bonds to the phosphate of the 4-phosphopantothenic acid portion of the cofactor; the amide nitrogen of Val44 forms a hydrogen bond to the amide carbonyl of the 4-phosphopantothenic acid portion of the cofactor; and finally the carboxyl oxygen of Thr42 forms a hydrogen bond to the nitrogen within the β-mercaptoethylamine portion of the cofactor. Comparison of the At1g77540 structure with that of At1g77540–CoA complex revealed a conformational change in the loop that spans residues 40–54 (Figure 6C). The largest displacement of the main chain occurs at Gly50, and the sidechain of Arg49 shows the most pronounced change in conformation. The electron density of the cofactor is well defined from the adenine ring to the Cβ of the β-alanine portion of the 4-phosphopantothenic acid. From that point, however, the cofactor appears to adopt multiple conformations. We modeled two alternative conformations of a CoA into the residual electron density. Interestingly, acetyl-CoA used to prepare complex with At1g77540 was apparently hydrolyzed (or enzymatically converted to CoA) in the process of crystal derivatization. We could not identify electron density corresponding to the acetyl group of acetyl-CoA. The sulfur atom of the thiol group of CoA resides close (3.1 Å) to the sulfur atom of absolutely conserved Cys76.
DISCUSSION
The X-ray and NMR solution structures of At1g77540 presented in this study clearly indicate that this protein adopts a fold similar to that previously observed in members of the GNAT superfamily. Primary sequence analysis of At1g77540 revealed that it belongs to the biochemically uncharacterized COG2388 family, which shows sequence relationship to the GNAT family of histone acetyltransferases (HATs) (30). We investigated this relationship in more detail and established that At1g77540 is indeed capable of binding acetyl-CoA as well as acetylating histones H3 and H4. The binding of acetyl-CoA is an absolute requirement for GNAT superfamily enzymes, and the preference for histones H3 and H4 is characteristic of eukaryotic GNAT family enzymes such as PCAF and GCN5 (38). The observed rate of histone acetylation by At1g77540 is, however, extremely low compared to other GNAT family HATs such as GCN5 (29) suggesting two possibilities: 1) histones are not the major substrate for At1g77540 and/or 2) At1g77540 requires other protein subunits or cofactors. In fact, nearly all known eukaryotic HATs function as large multiprotein complexes, which allows them to bind and recognize substrates more effectively than the catalytic subunit alone (39–41). At1g77540 is considerably shorter than other acetyltransferase domains, and its structure revealed what could be considered a “minimal” acetyltransferase fold. The main structural difference is that At1g77540 lacks the motif C features characteristic of GNATs. This “minimal” acetyltransferase thus may not provide the full range of interaction needed for specific binding and instead may require at least one partner to function efficiently.
There are two main general mechanisms of acetyl group transfer. One mechanism involves formation of an acetyl-enzyme intermediate and requires formation of two consecutive binary complexes to transfer the acetyl group to a substrate. The second mechanism requires formation of a ternary complex involving enzyme, acetyl-CoA and substrate. In the second case, the enzyme plays a role of a scaffold and provides residues that facilitate catalysis through direct attack of the substrate on the bound acetyl-CoA. Both types of mechanisms have been described for HATs: 1) yeast GCN5, a GNAT family enzyme, was shown to perform catalysis through a ternary complex, with Glu173 being responsible for deprotonation of histone lysine residues in order to form a nucleophile for direct attack of acetyl-CoA (29); 2) MYST family acetyltransferase Esa1 was shown to form an acetyl-enzyme intermediate, with Cys304 being acetylated. In addition, Glu338, acting as a general base, was shown to be required for catalysis (42).
Inspection of the structure of At1g77540 and its complex with CoA revealed three interesting features in the putative active site of this enzyme that may be relevant to other members of the COG2388 family. 1) The protein backbone connecting strand E and helix 4 adopts a unique conformation compared to other acetyltransferases, such as Hat1 and Esa1 (PDB # 1bob and PDB # 1mja, respectively) (Figure 7). This unique conformation results in closing of the cavity that usually accommodates the acetyl group of acetyl-CoA. 2) Instead, the strictly conserved Cys76 of the COG2388 family is located in this area (Figure 2). 3) A highly conserved His41 of the COG2388 family also maps to the putative active site (Figure 2). Whereas the proposed active site of At1g77540 exhibits obvious differences from those other known acetyltransferases, the loop connecting helices 2 and 3 of At1g77540 (highlighted by the black arrow in Figure 7) shares common structural features with other family members. This loop region, which is part of motif A, forms extensive hydrogen bonds with the pyrophosphate portion of CoA. Thus, it appears that local backbone conformations within this important CoA binding site are highly conserved.
Figure 7.
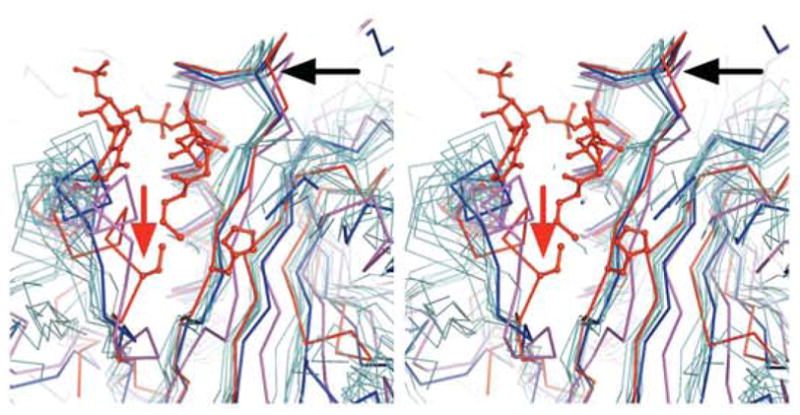
Structural alignment of the At1g77540/CoA complex with other acetyltransferases. The X-ray structure of the At1g77540-CoA complex is displayed in red. Other structures shown are: PDB # 1bob in blue, PDB # 1mja in magenta, and other members of the GNAT superfamily (PDB # 1bo4, 1cm0, 1i12, 1j4j, 1qsm, 1qsn, 1s5k, and 1tiq) in light blue. The sidechains of His41 and Cys76 are displayed as sticks. The black arrow denotes the approximate location of the loop between helices 2 and 3 of At1g77540, which contains the CoA binding motif. The red arrow denotes the location of the unique backbone conformation around Cys76.
We propose that At1g77540, and other COG23388 family enzymes, catalyze acetyl group transfer through formation of an acetyl-enzyme intermediate. According to our hypothesis, the strictly conserved Cys76 acts as a nucleophile that is acetylated in the first step of the reaction cycle (Figure 8). Cys76 is somewhat buried but may be deprotonated under physiological conditions. We found no residue in the vicinity of Cys76 that could be involved in direct deprotonation of this cysteine. The hydroxyl oxygen of the highly conserved Thr42 is located within 3.1 Å from the sulfur atom of Cys76 and could provide further stabilization of the side chain conformation of Cys76. His41 could be involved in the formation of the acetyl-enzyme intermediate. The imidazole nitrogen of His41 is located 3.5 Å from the sulfur of CoA in the At1g77540-CoA complex. On the basis of this geometry, we propose that the protonated form of His41 could be involved in the protonation of the acetyl-CoA on the sulfur atom, thus destabilizing the S–C thioester bond and making CoA a better leaving group (see Figure 8). Alternatively, at this stage of the reaction cycle, His41 could be involved in the stabilization of the oxyanion of the tetrahedral intermediate. More importantly, however, His41 may act as a general base during deprotonation of a substrate during the second transfer of the acetyl group. Depending on the identity of physiological substrate, His41 could activate primary amines in a way analogous to the histone lysine activation by Glu173 of GCN5, or it could activate hydroxyl groups as observed in the catalytic mechanism of chloramphenicol acetyltransferase (PDB # 3cla), where His195 acts as a general base to abstract a proton from the primary hydroxyl group of chloramphenicol (43). It is of interest to note that the catalytically active Cys304 of Esa1 is found in the location structurally equivalent to His41 of At1g77540, and not Cys76 of At1g77540. In addition, the location of Glu338 of Esa1 corresponds approximately to that of Ser75 of At1g77540, which is clearly not a conserved residue in COG2388. Similarly, the catalytically important Glu173 of yeast GCN5 corresponds structurally to Asp38 of At1g77540, another poorly conserved residue in COG2388. Taken together these comparisons suggest that At1g77540 likely represents a novel adaptation of the acetyltransferase fold tuned for a currently unknown substrate. Testing of the proposed mechanism will depend on identification of the true physiological substrate of At1g77540 and detailed kinetic studies to confirm or refute the presence of the acetylated intermediate and the roles of conserved Cys76 and His41. To date the only other COG2388 family member that has been structurally characterized is a putative N-acetyltransferase from Staphylococcus aureus (PDB # 1r57). The fold of this protein is identical to that of At1g77540, and the protein contains cysteine and histidine residues in locations analogous to Cys76 and His41 (Figure 2).
FIGURE 8.
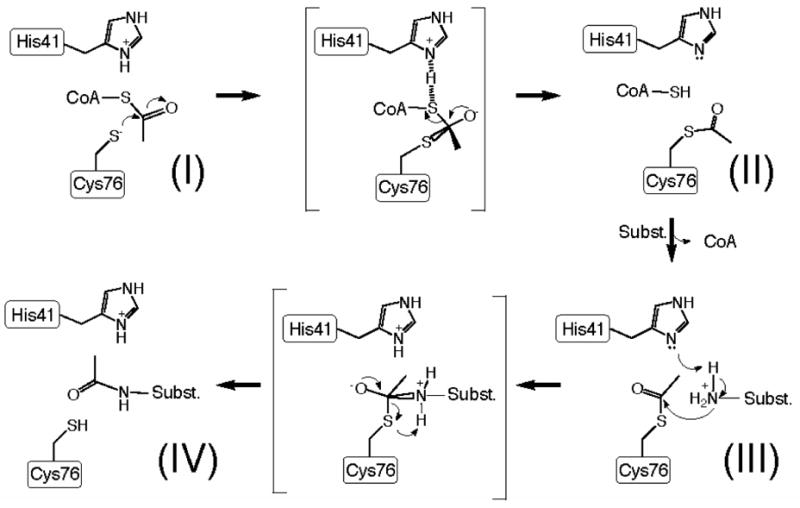
Proposed catalytic mechanism for substrate acetylation by At1g77540.
Inspection of the At1g77540-CoA complex reveals that the CoA in the binding pocket displays a kinked shape, with the β-mercaptoethylamine portion of the cofactor splayed between parallel β-strands D and E and with extensive hydrogen bonds to conserved residues in motif A. Similar binding patterns have been observed in other CoA complexes of GNAT superfamily enzymes (34, 44). The structure of the At1g77540-CoA complex can rationalize the conservation of Arg86 (or lysine at the same position) seen in multiple sequence alignment of the COG2833 family of proteins (Figure 2). The Arg86 residue is involved in electrostatic interactions with 3′-phosphate group of CoA, and thus stabilizes the binding of the cofactor. Similar stabilization was seen in recent structure of human GCN5 (PDB # 1z4r) with Lys624 providing the electrostatic stabilization.
In summary, we have solved the structure of A. thaliana At1g77540 protein by X-ray crystallography and solution NMR. Fold analysis confirmed that the protein is a new member of the GNAT superfamily with a minimal acetyltransferase fold. We have provided evidence that At1g77540 binds CoA in a manner similar to other members of the superfamily and established that At1g77540 is an acetyltransferase by enzymatic assays. Based on structural comparisons with other known GNAT family members we propose that At1g77540 acts through a unique mechanism that likely involves conserved His41 and the strictly conserved Cys76, which serves as nucleophile and undergoes acetylation; further studies will be needed to test this proposed mechanism. The identity of physiological substrate remains elusive and will be the object of further investigation.
Acknowledgments
We thank all members of the CESG team, including Todd Kimball, John Kunert, Nicholas Dillon, Rachel Schiesher, Juhyung Chin, Megan Riters, Andrew C. Olson, Jason M. Ellefson, Janet E. McCombs, Brendan T. Burns, Blake W. Buchan, Holalkere V. Geetha, Zhaohui Sun, Ip Kei Sam, Eldon L. Ulrich, Nathan S. Rosenberg, Janelle Warrick, Bryan Ramirez, Zsolt Zolnai, Peter T. Lee, Jianhua Zhang, David J. Aceti, Russell L. Wrobel, Ronnie O. Frederick, Hassan Sreenath, Frank C. Vojtik, Won Bae Jeon, Craig S. Newman, John Primm, Michael R. Sussman, Brian G. Fox. We thank Joe Brunzelle for facilitating data collection at LS-CAT at Advanced Photon Source Sector 32.
Footnotes
This work was supported by NIH grants P50 GM64598 and U54 GM074901 from the National Institute of General Medical Sciences (J.L.M., P.I). J.M.D. and C.E.B. acknowledge financial support from NIH grant GM059785. Use of the Advanced Photon Source and the Argonne National Laboratory Structural Biology Center beamlines, was supported by the U. S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38. We acknowledge LS-CAT for time at APS Sector 32. Use of the National Magnetic Resonance Facility at Madison was supported in part by NIH grant RR02301 from the National Center for Research Resources (NCRR). This work is also based upon research conducted at the Northeastern Collaborative Access Team (NE-CAT) beamlines of the Advanced Photon Source, supported by NIH grant RR15301 from the NCRR.
Atomic coordinates, along with structure factors for the X-ray structures and constraint lists for the NMR conformers, have been deposited in the Protein Data Bank, www.rcsb.org, under PDB # 2EVN (NMR structure of At1g77540), PDB # 1XMT (X-ray structure of At1g77540), and PDB # 2GDB (X-ray structure of At1g77540-CoA complex). Resonance assignments and primary NMR data have been deposited in BioMagResBank, www.bmrb.wisc.edu, under BMRB # 6338.
Abbreviations: APS Argonne National Laboratory Advanced Photon Source; At1g77540, the protein product produced from Arabidopsis thaliana gene At1g77540.1; CESG, Center for Eukaryotic Structural Genomics; CoA, coenzyme A, GNAT, GCN5-related N-acetyltransferases family; HATs, histone acetyltransferases; r.m.s.d., root-mean-square deviation.
References
- 1.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 2.Vetting MW, de Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7(Suppl):957–959. doi: 10.1038/80734. [DOI] [PubMed] [Google Scholar]
- 4.Thao S, Zhao Q, Kimball T, Steffen E, Blommel PG, Riters M, Newman CS, Fox BG, Wrobel RL. Results from high-throughput DNA cloning of Arabidopsis thaliana target genes using site-specific recombination. J Struct Funct Genomics. 2004;5:267–276. doi: 10.1007/s10969-004-7148-4. [DOI] [PubMed] [Google Scholar]
- 5.Sreenath HK, Bingman CA, Buchan BW, Seder KD, Burns BT, Geetha HV, Jeon WB, Vojtik FC, Aceti DJ, Frederick RO, Phillips GN, Jr, Fox BG. Protocols for production of selenomethionine-labeled proteins in 2-L polyethylene terephthalate bottles using auto-induction medium. Protein Expression Purif. 2005;40:256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 6.Jeon WB, Aceti DJ, Bingman CA, Vojtik FC, Olson AC, Ellefson JM, McCombs JE, Sreenath HK, Blommel PG, Seder KD, Burns BT, Geetha HV, Harms AC, Sabat G, Sussman MR, Fox BG, Phillips GN., Jr High-throughput Purification and Quality Assurance of Arabidopsis thaliana Proteins for Eukaryotic Structural Genomics. J Struct Funct Genomics. 2005;6:143–147. doi: 10.1007/s10969-005-1908-7. [DOI] [PubMed] [Google Scholar]
- 7.Zolnai Z, Lee PT, Li J, Chapman MR, Newman CS, Phillips GN, Jr, Rayment I, Ulrich EL, Volkman BF, Markley JL. Project management system for structural and functional proteomics: Sesame. J Struct Funct Genomics. 2003;4:11–23. doi: 10.1023/a:1024684404761. [DOI] [PubMed] [Google Scholar]
- 8.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Tyler RC, Sreenath HK, Singh S, Aceti DJ, Bingman CA, Markley JL, Fox BG. Auto-induction medium for the production of [U-15N]- and [U-13C, U-15N]-labeled proteins for NMR screening and structure determination. Protein Expression Purif. 2005;40:268–278. doi: 10.1016/j.pep.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 10.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 11.Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Crystallogr D. 2003;59:1966–1973. doi: 10.1107/s0907444903018043. [DOI] [PubMed] [Google Scholar]
- 12.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallogr A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 13.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 14.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 15.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 16.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 18.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 19.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- 20.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMR Pipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 24.Eghbalnia HR, Bahrami A, Wang L, Assadi A, Markley JL. Probabilistic Identification of Spin Systems and their Assignments including Coil-Helix Inference as Output (PISTACHIO) J Biomol NMR. 2005;32:219–233. doi: 10.1007/s10858-005-7944-6. [DOI] [PubMed] [Google Scholar]
- 25.Nilges M, O’Donoghue SI. Ambiguous NOEs and automated NOE assignment. Progress in Nuclear Magnetic Resonance Spectroscopy. 1998;32:107–139. [Google Scholar]
- 26.Linge JP, Nilges M. Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J Biomol NMR. 1999;13:51–59. doi: 10.1023/a:1008365802830. [DOI] [PubMed] [Google Scholar]
- 27.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 28.Seavey BR, Farr EA, Westler WM, Markley JL. A relational database for sequence-specific protein NMR data. J Biomol NMR. 1991;1:217–236. doi: 10.1007/BF01875516. [DOI] [PubMed] [Google Scholar]
- 29.Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 30.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005;33:D192–196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 32.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 33.Madej T, Gibrat JF, Bryant SH. Threading a database of protein cores. Proteins. 1995;23:356–369. doi: 10.1002/prot.340230309. [DOI] [PubMed] [Google Scholar]
- 34.Dutnall RN, Tafrov ST, Sternglanz R, Ramakrishnan V. Structure of the histone acetyltransferase Hat1: a paradigm for the GCN5-related N-acetyltransferase superfamily. Cell. 1998;94:427–438. doi: 10.1016/s0092-8674(00)81584-6. [DOI] [PubMed] [Google Scholar]
- 35.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 37.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell. 1998;94:439–449. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 38.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 39.Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Cote J. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev. 2003;17:1415–1428. doi: 10.1101/gad.1056603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doyon Y, Cote J. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev. 2004;14:147–154. doi: 10.1016/j.gde.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Selleck W, Fortin I, Sermwittayawong D, Cote J, Tan S. The Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase complex requires the Enhancer of Polycomb A domain and chromodomain to acetylate nucleosomes. Mol Cell Biol. 2005;25:5535–5542. doi: 10.1128/MCB.25.13.5535-5542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–869. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 43.Leslie AG, Moody PC, Shaw WV. Structure of chloramphenicol acetyltransferase at 1.75-Å resolution. Proc Natl Acad Sci U S A. 1988;85:4133–4137. doi: 10.1073/pnas.85.12.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Y, Barlev NA, Haley RH, Berger SL, Marmorstein R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol Cell. 2000;6:1195–11205. doi: 10.1016/s1097-2765(00)00116-7. [DOI] [PubMed] [Google Scholar]
- 45.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]