

Abstract
Summary: The heat shock response (HSR) is a homeostatic response that maintains the proper protein-folding environment in the cell. This response is universal, and many of its components are well conserved from bacteria to humans. In this review, we focus on the regulation of one of the most well-characterized HSRs, that of Escherichia coli. We show that even for this simple model organism, we still do not fully understand the central component of heat shock regulation, a chaperone-mediated negative feedback loop. In addition, we review other components that contribute to the regulation of the HSR in E. coli and discuss how these additional components contribute to regulation. Finally, we discuss recent genomic experiments that reveal additional functional aspects of the HSR.
INTRODUCTION
The heat shock response (HSR) is classically defined as the cellular response to temperature increase. A major component of this response is the upregulation of a set of proteins termed heat shock proteins (hsp's). These proteins are usually regulated by a single transcription factor, for example, the σ32 transcription factor in Escherichia coli and heat shock factor (HSF) in eukaryotic cells. Some hsp's, notably chaperones, which help proteins fold, and proteases, which degrade unfolded proteins, are a conserved part of the response from bacteria to humans. The rapid upregulation of chaperones and proteases during the HSR restores an appropriate protein-folding environment in the cell, suggesting that maintaining protein-folding homeostasis is a primary function of the σ32/HSF-mediated HSR. Consistent with this idea, many other treatments that destabilize folded proteins or make it more difficult for nascent proteins to fold also activate this response.
In this review, we discuss the regulation and function of the E. coli σ32-mediated HSR. We show that this response is subject to complex regulation, which is currently not completely understood. In addition, we discuss the broad reach of the σ32 regulon, which encodes functions that alter multiple cellular processes to permit increased survival in response to high temperature.
A PHYSIOLOGICAL DESCRIPTION OF THE HSR
The σ32-mediated HSR has been characterized by examining the behavior of the response after both upshift and downshift in temperature. Following upshift to temperatures of ≥37°C but within the growth range of the organism, the synthesis of hsp's first increases rapidly during the induction phase of the response, then declines during the adaptation phase of the response, and finally achieves a new steady-state level characteristic of the particular high temperature (Fig. 1A) (53). The initial rapid increase in synthesis allows hsp's to reach the level characteristic of the new temperature rapidly. Conversely, following temperature downshift, hsp synthesis abruptly decreases about 20-fold and then gradually increases over the next several doublings until it reaches the rate characteristic of 30°C (Fig. 1B) (52). Excess hsp's are diluted out as cells grow; it is believed that the normal rate of hsp synthesis resumes when the cellular level of hsp's approximates that characteristic of growth at 30°C.
FIG. 1.

Activation and repression of the HSR during temperature upshift and downshift. (A) Activation of the HSR during a temperature shift from 30 to 42°C reveals three distinct phases: induction, adaptation, and steady state. (B) Repression of the HSR during a temperature shift from 42° to 30°C. The relative σ32 activities measured by HSP synthesis are shown by the solid lines; relative σ32 levels measured by Western blotting analysis are shown by the dotted lines.
Three regulatory loops control the output by altering the level and activity of σ32 (Fig. 2) (reviewed in reference 67). First, translation of σ32 increases at high temperature. Second, σ32 stability is controlled: σ32 is rapidly degraded during steady-state growth at both low and high temperatures but transiently stabilized after shift to high temperature. Finally, σ32 activity is negatively regulated. Activity control is believed to adjust σ32-mediated transcription to a rate appropriate for the level of unfolded proteins present in the cell, leading to the transient inactivation of σ32 when excess hsp's are present. During the induction phase of the HSR, increased translation and transient stabilization of σ32 result in a rapid increase in its level (Fig. 1A), while the high pool of unfolded proteins removes the negative regulation of σ32 activity. Together, these mechanisms account for the rapid increase in hsp synthesis during the induction phase. During the adaptation phase, activity control is believed to mediate the decline in hsp synthesis that precedes the decline in σ32 levels (Fig. 1A). Likewise, following temperature downshift, activity control is believed to mediate the decline in hsp synthesis that precedes the decline in σ32 levels (Fig. 1B). The effect of each regulatory loop on response performance has been modeled; these data will be discussed later in the review.
FIG. 2.

Wiring diagram of σ32 regulation. There are three primary modes of regulation as follows: (i) excess free DnaK/J and GroEL/S chaperones directly bind to and inactivate σ32; (ii) the FtsH protease degrades σ32, with chaperones participating in this process; and (iii) temperature directly controls the rate of σ32 translation. Misfolded proteins titrate chaperones from these regulatory roles, allowing active σ32 to increase the synthesis of chaperones and proteases during conditions where they are needed.
TRANSLATIONAL CONTROL OF σ32
Early studies demonstrated that the rate of translation of σ32 was controlled by temperature: σ32 synthesis was 5- to 10-fold higher at 42°C than at 30°C (14, 39, 40, 53). A structural transition in rpoH mRNA encoding σ32 mediates this control: at low temperature, base pairing within rpoH mRNA occludes the Shine-Dalgarno sequence and the translation start point of σ32, resulting in inefficient initiation of translation; at high temperature, this inhibitory structure is melted so that the translation of σ32 increases (35, 37). The evidence for this view is as follows: (i) destabilizing the predicted inhibitory structure by single base changes increases the translation of σ32 at low temperature, whereas stabilizing the inhibitory structure with single base changes decreases the translation of σ32 at all temperatures; and (ii) temperature regulation and the effects of stabilizing and destabilizing mutations can be reproduced in vitro by assaying ribosome binding to rpoH mRNA. These experiments suggest that translation control responds directly to changes in temperature rather than sensing the cellular folding environments that result from the temperature changes. It is not known whether additional factors or stresses alter the performance of this switch in vivo.
REGULATION OF σ32 ACTIVITY
Control of σ32 activity was initially inferred from the fact that under conditions where there is transiently more σ32 than necessary, expression of hsp's is lower than expected from the amount of σ32 present in the cell (52). Thus, activity regulation is observed during the adaptation phase of temperature upshift (Fig. 1A), after temperature downshift (Fig. 1B), and when σ32 is artificially overexpressed. The “unfolded protein titration model” has been proposed to explain activity control. According to this model, σ32 samples chaperone activity to sense the amount of unfolded proteins in the cell: when chaperone levels are abundant relative to unfolded proteins, the chaperones would feedback inhibit σ32 and therefore negatively regulate further chaperone production. Consistent with this idea, the overexpression of either the DnaK/J or the GroEL/S chaperone machine inactivates σ32; conversely, depleting either chaperone machine leads to the accumulation of active σ32 (16, 58). The DnaK/J chaperone machine, consisting of the Hsp70 homologue DnaK and its Hsp40 cochaperone DnaJ, and the GroEL/S chaperone machine, consisting of the Hsp60 homologue GroEL and its Hsp10 cochaperone GroES, represent the two major cytoplasmic chaperones in Escherichia coli. The strongest evidence supporting the “unfolded protein titration model” is that the expression of unstable proteins that titrate either DnaK/J or GroEL/S induces the HSR (16, 58). This suggests that σ32 is not responding to the total level of chaperones in the cell but rather is responding to the ratio of chaperone relative to those of its unfolded protein substrates. This control circuit would allow the cell to continuously monitor its protein-folding state to ensure that the expression of chaperones was appropriate for the unfolded substrate load. Consistent with this model, chaperones do not participate in stable interactions with their substrates; rather, they engage in cycles of substrate binding and release that are driven by the ATPase activity of the chaperones.
The molecular mechanism of the chaperone-mediated inactivation of σ32 is not completely settled. The prevalent model is that the chaperones effectively act as anti-sigma factors and simply compete with RNA polymerase for binding to σ32. In support of this idea, both DnaK/J and GroEL/S bind to σ32 in vitro, and the addition of either DnaK/J or GroEL/S to an in vitro transcription reaction mixture containing purified σ32 and RNA polymerase leads to a decrease in σ32-dependent transcription (10, 11, 16, 32). However, there are accumulating hints that the inactivation mechanism may be more complex than simple competition. First, σ32 binds to GroEL/S with an affinity ∼1,000× lower than that with which it binds to RNA polymerase. Yet, a small excess of GroEL/S over RNA polymerase (∼5-fold) efficiently inactivates σ32 in vitro, which cannot be explained by a competitive binding model (16). Second, although in vitro binding studies indicate that GroEL/S binds ∼100-fold more weakly to σ32 than does DnaK/J, GroEL/S is at least as efficient as DnaK/J in mediating inactivation both in vivo and in vitro (16). Finally, point mutants have been identified in σ32 that are resistant to inactivation by both GroEL/S and DnaK/J in vivo, but these mutants are inactivated indistinguishably from the wild type in the current in vitro inactivation assay (these mutants are discussed further in the section on σ32 structure/function). Together, these observations raise the possibility that an additional component(s) may be involved in inactivation.
Thus far, we have considered only the action of the major Hsp40 family member, DnaJ, in mediating activity control. However, E. coli has other Hsp40s, and one of them, CpbA, is known to be able to mediate activity control in vivo in collaboration with DnaK (60, 61). It is not known to what extent σ32 is regulated by DnaK/J versus DnaK/CbpA. Moreover, CbpA has not been examined in vitro in regard to σ32 binding or inactivation. Also, CbpA activity can be modulated in vivo by the accessory factor CbpM (5). It will be interesting to determine the conditions under which CbpA and CbpM are important in mediating activity control.
There is another potential contribution to the activity control mechanism just described. The DnaK/J chaperone machine requires the GrpE nucleotide exchange factor to exchange ADP for ATP, therefore allowing substrate release and a new round of substrate binding. There is evidence that GrpE can act as a thermosensor (reviewed in reference 65). At high temperatures, GrpE activity decreases, leading to a slower ATPase cycle, which in turn leads to DnaK/J acting more like holdase than a foldase. Whether this alteration in the functional properties of GrpE affects the activity control of σ32 has not been investigated.
Finally, the cell can adapt to long-term imbalances in the ratio of chaperones to unfolded proteins. Whereas the overexpression of either DnaK/J or GroEL/S results in an immediate inhibition in σ32 activity, following long-term (20-h) overexpression of chaperones, the σ32 activity level is upregulated to approximately that of wild-type cells (17). Adaptation is accomplished without the downregulation of the overexpressed chaperones. The players in this regulatory loop are currently unknown.
DEGRADATION CONTROL OF σ32
The degradation control of σ32 is complex. σ32 is degraded rapidly during steady-state growth, exhibiting a half-life of ∼1 min at low temperature (30°C) and an even higher rate of degradation (half-life of ∼20 s) at high temperature (42°C) (27, 36). In addition, immediately after a shift to high temperature, this normally unstable protein is transiently stabilized for a 5- to 10-min period (53). Currently, some but not all features of this response are understood.
A search for the factors involved in the degradation of σ32 indicated that FtsH (HflB), an ATP-dependent protease localized to the inner membrane, is the major protease that degrades σ32. σ32 is almost completely stable in cells lacking FtsH, whereas single deletions of other cytoplasmic ATP-dependent proteases have little or no effect on σ32 stability (20, 26, 57). In addition, the DnaK/J and GroEL/S chaperone machines are implicated in σ32 degradation, as depleting either chaperone machine or mutationally inactivating DnaK or DnaJ stabilizes σ32 (16, 51, 58). The involvement of chaperones in σ32 degradation means that the “unfolded protein titration model” could explain the transient stabilization of σ32 after temperature upshift as well as the activity control of σ32. The increased prevalence of unfolded proteins after a shift to high temperature would titrate chaperones away from their role in degradation so that σ32 would be transiently stabilized. If so, this would allow the rate of degradation to directly monitor the folding status of the cell. Additionally, the increased prevalence of FtsH substrates after temperature upshift could titrate FtsH from degrading σ32, resulting in its transient stabilization, thereby tying the degradation rate of σ32 to substrate flux through FtsH. However there are conflicting data about whether FtsH is a limiting component in the degradation reaction (20, 55).
Several groups have investigated the thermal behavior of σ32. Both protease sensitivity and hydrogen-deuterium exchange experiments coupled with mass spectrometry indicate that σ32 becomes more unstructured at high temperatures (47). This may partially explain the extremely rapid degradation of σ32 at high temperatures. In addition, FtsH itself has increased activity at higher temperatures (19, 27).
Importantly, the degradation system has not been completely reconstituted in vitro. σ32 is degraded very rapidly in vivo, but degradation by FtsH in vitro is very slow (20, 57). Moreover, the DnaK and DnaJ chaperones do not facilitate the degradation of σ32 in vitro (3). Investigation of the properties of FtsH revealed that it has a very poor unfoldase activity both in vivo and in vitro, so that it essentially waits for proteins to spontaneously unfold before degrading them (19). The low rate of σ32 degradation in vitro could reflect the time required for the unfolding of σ32. Intriguingly, FtsH is a member of the AAA family of proteins, many of which utilize adaptor proteins to modulate their activity (reviewed in reference 7). For example, the degradation of σS by the AAA+ protease ClpXP requires the RssB adaptor protein (69). The putative FtsH adaptor protein(s) missing from the in vitro system could provide unfoldase activity and/or recruit chaperones.
The role of chaperones in σ32 degradation is not settled. One prevalent model is that chaperones facilitate σ32 degradation by competing with RNA polymerase for binding to σ32. In support of this model, RNA polymerase prevents the degradation of σ32 by FtsH in vitro, and the addition of chaperones reverses this effect (3). However, it has recently been shown that chaperones are still required for the in vivo degradation of σ32 mutants that are defective in RNA polymerase binding (54).
σ32 STRUCTURE/FUNCTION
σ32 is a member of the bacterium-specific σ transcription factor family (reviewed in reference 15). All σ's contain binding determinants both for RNA polymerase and for promoter DNA. Binding of a σ to RNA polymerase induces changes both in the σ and in RNA polymerase; the resultant holoenzyme is competent to bind to promoters specified by the particular σ factor utilized. Bacteria generally contain a single housekeeping σ factor and several alternative σ's which mediate responses to altered environmental conditions. σ's contain between two to four domains, depending on the particular group to which they belong. Housekeeping σ's are the most complex and contain four domains; σ32, a member of the group 3 σ's, contains domains 2,3, and 4 (Fig. 3). Several domains contain recognition determinants for RNA polymerase binding and for promoter recognition. Domain 2 recognizes the −10 region of the promoter and carries the major RNA polymerase recognition determinants, whereas domain 4 recognizes the −35 region of the promoter. There is a reasonable amount of knowledge about the RNA polymerase and promoter recognition determinants in σ32 both as a result of direct studies on σ32 and by extrapolation from studies on other σ's.
FIG. 3.

The domain structure of σ32. A schematic map reveals the domain structure and conserved regions of σ32. Note that domains are divided into subdomains as follows: domain 2 is comprised of subdomains 1.2 to 2.4 and encompasses amino acids 16 to 126, domain 3 is comprised of subdomains 3.1 and 3.2 and encompasses amino acids 127 to 177, and domain 4 is comprised of subdomains 4.1 and 4.2 and encompasses amino acids 213 to 280. The RpoH box is comprised of amino acids 132 to 141. Regions that bind RNA polymerase and promoter DNA are shown below the schematic; features specific to σ32 are indicated above the schematic. “Activity/stability mutants” marks the position of mutations in σ32 that affect the stability and/or activity of σ32.
In addition to carrying out the functions common to all σ's, σ32 must contain determinants that allow it to bind to several chaperones and to the FtsH protease, so that its activity and stability can be properly regulated. However, it has proven surprisingly difficult to identify mutations that are specifically altered in its regulatory determinants. One reason for this is that the multiple regulatory loops tend to obscure the true phenotype of such mutations. For example, mutations in σ32 that eliminate the binding site for the FtsH protease would result in the accumulation of high levels of σ32 but would not significantly increase σ32 activity, because activity control would inactivate excess σ32.
The initial search for a region of σ32 specialized to carry out regulatory functions focused on the RpoH box (part of the previously described region C) (38, 41). As this region is unique to σ32 and its orthologues in other bacteria, it was an excellent candidate for a region devoted to functions unique to σ32. The RpoH box spans amino acids 122 to 144 and is located at the N terminus of domain 3 (Fig. 3). Two peptides from within the RpoH box region bind DnaK, but it is unlikely that these are used as DnaK binding determinants in intact σ32 because mutating these sites did not lead to defects in σ32 regulation (1, 33). A frameshift mutation spanning the RpoH box stabilized σ32 (38); later studies showed that this peptide is a substrate for FtsH (1), making this a candidate for the FtsH recognition sequence. However, analysis of the various rpoH paralogues from Bradyrhizobium japonicum, which contains both stable and unstable σ32s, indicated that differences in degradation control did not map to the RpoH box (62). Thus, there is no strong evidence that the RpoH box is involved in either chaperone or protease binding to σ32. At present, the only known function of the RpoH box is binding to RNA polymerase (1, 25).
A great deal of attention has been focused on mapping the degradation determinants in σ32. Motivated by the finding that ATP-dependent proteases often use C-terminal recognition determinants, the role of the C terminus of σ32 in degradation control was investigated. Initial studies showed that C-terminal truncations of 15 or 20 amino acids led to the stabilization of σ32 both in vivo and in vitro but did not affect DnaK/J binding (3). However, these truncations had additional vector-encoded amino acids added to their C termini. When C-terminal truncations of 5, 11, 15, or 21 amino acids without additional vector sequences were analyzed, these proteins exhibited the same high rate of degradation in vivo as wild-type σ32 (56). Moreover, FtsH does not efficiently degrade a peptide derived from the C terminus of σ32. Another approach to mapping the degradation control region utilized chimeras between E. coli σ32 and its B. japonicum orthologue, rpoH1 (2). The σ32 encoded by rpoH1 is 10 times more stable than E. coli σ32, although the two proteins are 40% identical. This work suggests that the main degradation tag lies somewhere between amino acids 36 and 134.
Several groups have used forward genetic screens to search for σ32 mutants with alterations in either activity or stability (21, 45, 66). Importantly, these diverse screens with different endpoints have converged on a small region within domain 2.1 of σ32. In one screen, the degradation control phenotype was completely uncoupled from activity control by assaying for the expression of a chimeric adenylyl cyclase whose N and C termini were separated by σ32 (45). Because σ32 is unstable, the chimeric protein is degraded and adenylyl cyclase activity is low; mutations in σ32 resulting in a degradation defect will have higher adenylyl cyclase activity. Importantly, screens for σ32 mutants defective in activity regulation found similar affected residues (21, 66). Residues identified in all of the forward genetic screens are shown in Table 1. Analysis of the mutant most defective in activity control (σ32 I54N) indicated that it was almost unaffected by the overexpression of either DnaK/J or GroEL/S and simultaneously completely defective in degradation control (66). Activity and degradation control are linked because both processes require chaperone binding. Yet, this mutant showed essentially normal binding to DnaK, DnaJ, and GroEL and was also almost normal in its binding to RNA polymerase. This regulatory region may affect a step downstream of chaperone binding that is important for both processes. For example, it may orchestrate a conformational change or bind an unknown factor. Although the precise defect of these mutants is unknown, the analysis performed thus far indicates that these mutants identify a critical regulatory region within σ32 that is important for both activity and stability control.
TABLE 1.
Mutations that affect the stability and/or activity of σ32
| Mutation | Alteration(s) | Reference(s) with identification |
|---|---|---|
| L47A | Stability | 21 |
| L47Q | Stability | 21 and 45 |
| L47Q-L55Q | Activity, stability | 21 |
| A50D | Activity, stability | 66 |
| A50S | Stability | 21 |
| A50T | Stability | 21 |
| A50V | Stability | 45 |
| K51E | Activity, stability | 21 and 66 |
| I54A | Activity, stability | 21 |
| I54F | Stability | 45 |
| I54N | Activity, stability | 66 |
| I54T | Activity, stability | 21, 45, and 66 |
| L55Q | Stability | 21 |
| R91H | Activity, stability | 66 |
| R91P | Activity, stability | 66 |
| Δ49-52 | Activity, stability | 66 |
MODELING REGULATION OF THE HSR
The σ32-mediated HSR is an attractive candidate for mathematical modeling because it is well studied experimentally and known to be subject to complex regulation. Several groups have modeled the response (8, 9, 31, 49). Perhaps the most interesting results were obtained by considering the HSR in the context of control engineering, a discipline that uses modular decomposition to make systems tractable for analysis. In this representation, temperature-regulated translation is considered to be a feed-forward module allowing the system to respond to change in temperature before cellular processes are altered (Fig. 4). Additionally, two feedback loops, one mediating activity control and a second mediating degradation control, report on cellular conditions, allowing a homeostatic response. To examine the function of each of these modules, the response properties of “virtual mutants” that had various combinations of these modules were modeled. This analysis revealed that regulation is not redundant; instead, each module contributes different features to the response. The simplest system is one in which control is exerted solely by a direct sensor of temperature (temperature-regulated translation); as σ32 increases, chaperones and proteases increase concomitantly. This system can achieve any output desired but does not respond to the internal state of the cell because it contains no feedback loops. Such a system is inefficient because it utilizes many chaperones to accomplish folding at elevated temperatures even if the level of unfolded proteins is low, and it is also very sensitive to parameter variation—the system changes in concert with changes in parameters. The addition of activity control improves the efficiency of the system and makes it less sensitive to parameter variation. The further addition of degradation control improves the kinetics of the response, increases its efficiency, and reduces cell-to cell variation. Analyses of this type rationalize the complexity of biological control mechanisms. The availability of such a detailed model begs for experimental tests of these predictions. Should the experimental tests fail to validate the model, this would suggest that additional control features are present, thereby motivating a new round of investigation of the response. Likewise, the identification of additional control mechanisms experimentally would motivate the development of a new model.
FIG. 4.
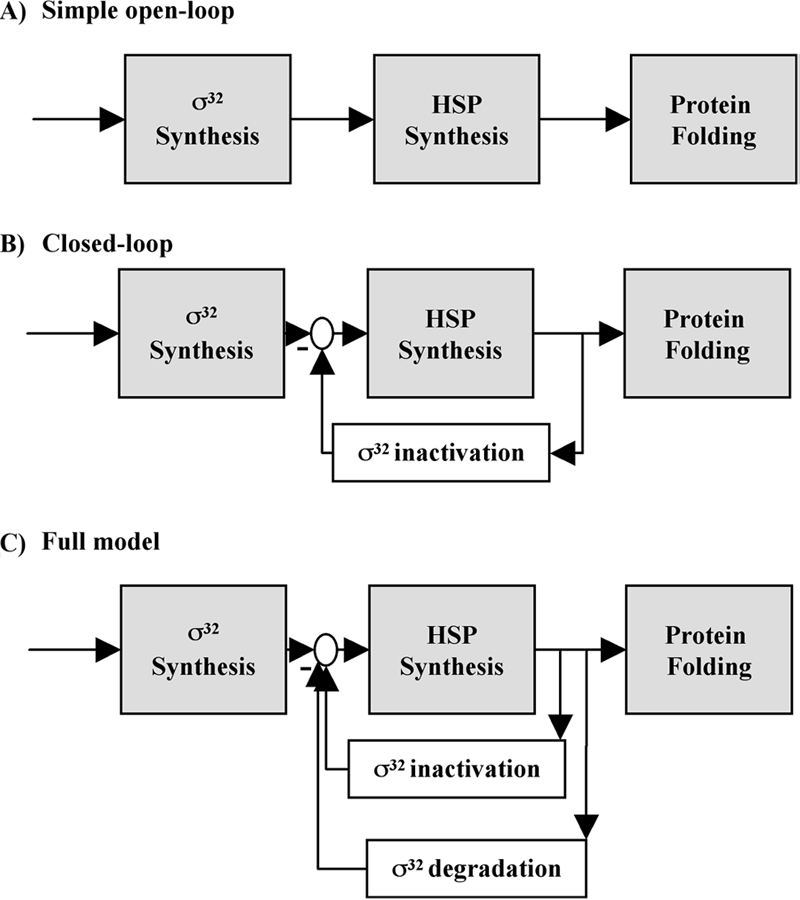
Modeling the HSR. (A) A simple open-loop design containing only a feed-forward element that senses temperature. (B) A closed-loop design with feed-forward and inactivation loops. (C) A full model containing feed-forward, inactivation, and degradation loops. (Reprinted from reference 9 with permission of the publisher. Copyright 2005 National Academy of Sciences, U.S.A.)
THE FUNCTIONS OF THE σ32 REGULON
The σ32-mediated HSR is the most immediate response of E. coli and related organisms to temperature stress. Therefore, identifying the functions encoded by this regulon will allow us to determine the cellular alterations that permit adaptation to this ubiquitous stress. Recently, the functions encoded by the regulon have been identified either by using whole-genome expression analysis to identify RNAs that increase after overexpression of σ32 and transfer to high temperature (44, 68) or by examining σ32 holoenzyme binding to DNA after temperature upshift by use of chromatin immunoprecipitation coupled with microarray analysis (63). The cellular location and functions of proteins known or likely to be part of the σ32 regulon are presented in Table 2.
TABLE 2.
Localization and functional classification of σ32 regulon productsa
| Location or functional category (no. of products) | Regulon products |
|---|---|
| Location | |
| Periplasm (8) | CreAc, DsbCc, YceIc, YciMb, YehRe, YehZe, YgcIe, YibGe |
| Inner membrane (27) | CreCc, CutEc, CycAb, FtsHd, FxsAb,c,e, GntYc, HflXd, HflKd, HflCd, HtpXd, LipBc, LspAc, MacBb, MenAd, PgpAc, PhoQc, SdaCe, YafUe, YbeXc, YbeZb,c,e, YceJc, YcjFb,c, YciSb,c,e, YdgRe, YghJe, YiaAe, YtfLe |
| Cytoplasm (88) | ClpBd, ClpPd, ClpXd, CreBc, Crre, DnaJd, DnaKd, FkpBc, FimBe, FolPd, GapAd, GlnSc,e, GntXc,e, GroELSd, GrpEd, HepAc,e, Hfqc, HolCc,e, Hsp33c, HslUd, HslVd, HtpGd, IbpAd, IbpBd, IleSc,e, IspHc, LdhAb,e, Lond, MiaAd, Mfdb,e, Mlcd, MutLd, MutMb,c, NarPc,e, NrdHe, NusBc, PhoPc,e, PrlCb,c,e, PyrFc, RecAc, RecJc, RdgBc, RibEc, RluAb, RpmEe, RpoDd, RpsLe, RrmJd, rrnBd, SdaAc,e, ThiLc, TopAd, TreRe, TyrRc, ValSc, XerDc, YadFc,e, YafDc, YafEb,c, YbbNb,e, YbeDb,c,e, YbeYc, YbjXe, YccEe, YccVb,e, YcePb,c,e, YciHc, YcjXb,c,e, YdaMb, YdeOe, YdhQb,c,e, YeaDd, YfjNc, YfiAe, YgaDc, YgbFc, YggWc, YhdNc,e, YhiQb,c, YibAb,e, YjhIe, YjiTe, YnfKd, YrdAb,e, YrfHb,c, YrfGc,e, XapRe, ZntRc |
| Unknown (3) | yi81_1d, yi82_1d, YpjMe |
| Functional category | |
| Metabolism (22) | CutEc, FolPd, GapAd, IspHc, LdhAb,e, LipBc, LspAc, MenAd, NrdHe, PgpAc, PyrFc, RibEc, SdaAc,e, ThiLc, YadFc, YafEb,c, YceJc, YdaMb, YggWc, YibAb,e, YrdAb,e, YrfGc,e |
| Chaperone/folding catalysts (12) | ClpBd, DnaJd, DnaKd, DsbCc, FkpBc, GroELSd, GrpEd, Hsp33c, HtpGd, IbpAd, IbpBd, YbbNb,e |
| Protein degradation (11) | ClpPd, ClpXd, FtsHd, HflXd, HflKd, HflCd, HslUd, HslVd, HtpXd, Lond, PrlCb,c,e |
| DNA modification (9) | HolCc,e, Mfdb,e, MutLd, MutMb,c, RecAc, RecJc, RdgBc, TopAd, XerDc |
| RNA state (3) | Hfqd, RluAb, YfjNc |
| Transcription regulators (18) | CreBc, CreCc, CycAb, FimBe, HepAc,e, MacBb, Mlcd, NarPc,e, NusBc, PhoPc,e, PhoQc, RpoDd, TreRe, TyrRc, XapRe, YdeOe, YjhIe, ZntRc |
| Translation/tRNA (11) | GlnSc,e, IleSc,e, MiaAd, RpmEe, RpsLe, RrmJd, rrnBc, ValSc, YciHc, YfiAe, YrfHb,c |
| Transporter (7) | Crrc, GntXc,e, SdaCe, YbeXc, YbeZb,c,e, YdgRe, YehZe |
| Miscellaneous (6) | FxsAb,c,e, GntYc, YccVb,e, YhiQb,c, yi81_1d, yi82_1d |
| Unknown function (27) | CreAc, YafDc,e, YafUe, YbeDb,c,e, YbeYc, YbjXe, YccEe, YceIc,YcePb,c,e, YciMb,c, YciSb,c,e, YcjFb,c, YcjXb,c,e, YdhQb,c,e, YeaDd, YehRe, YgaDc, YgbFc, YgcIe, YghJe, YhdNc,e, YiaAe, YibGe, YjiTe, YnfKd, YpjMe, YtlLe |
The predicted functions (http://ecocyc.org) and locations (46) were obtained for all identified σ32 regulon products. Note that rrnB is an RNA. Underlined proteins are annotated as “membrane”; however, it is assumed that they are located in the inner membrane.
Identified in reference 67.
Identified in reference 43. Only regulon products encoded on the chromosome and with a confirmed σ32-dependent promoter are listed.
Previously known; referred to in reference 43.
Identified in reference 62.
The earliest identified members of the regulon were chaperones, suggesting that maintaining protein homeostasis was an important role of the regulon (reviewed in references 12 and 13). The centrality of this function of the HSR is evidenced by the fact that these functions are under heat shock control in organisms as diverse as bacteria and humans. However, analysis of regulon members reveals a new dimension in protein homeostasis, complex cellular proteins containing moieties including iron-sulfur clusters, lipoyl modifications, and cofactors. It is perhaps not surprising that many members of the regulon are devoted to the homeostasis of complex proteins, as these proteins may be particularly at risk after a switch to high temperature, and regeneration pathways for some of these proteins have not been identified. The regulon encodes at least two iron-sulfur cluster proteins; it is possible that the synthesis of these proteins compensates for their destruction (reviewed in reference 23). Additionally, the regulon encodes proteins involved in lipoyl biosynthesis, iron-sulfur assembly (GntY, a homologue of IscA), and cofactor biosynthesis, thus ensuring a sufficient flux of the building blocks for complex proteins.
In addition to protecting proteins from destruction, functions encoded by the σ32 regulon also protect other macromolecules and cellular processes (Fig. 5A). A number of regulon functions protect cellular DNA; the regulon includes Mfd, which recruits repair machinery to DNA lesions, as well as enzymes involved in three different pathways that maintain genomic integrity (mismatch repair, excision repair, and recombination) (48). Enzymes for modifying rRNA (23S methylation) and tRNA (Δ3-isopentyl diphosphate added adjacent to the anticodon) are encoded by the regulon; these modifications are believed to be important for growth at high temperature (4, 59). Several RNA polymerase binding proteins (HepA, TopA, and NusB) believed to be important for alleviating the effects of supercoiling on transcription and two proteins implicated in ribosome protection and recycling (YfrH [Hsp15] and YfiA) are regulon members (6, 29, 30, 50). Taken together, this brief survey indicates that σ32 regulon members protect DNA and RNA in addition to proteins from the deleterious effects of excess heat and also modify the transcription, translation, and repair machinery to enable stress adaptation.
FIG. 5.

Functions of the HSR. The induction of σ32 and protein products of the target heat shock (hs) genes are shown in a model of the E. coli cell, illustrating the compartmentalization of the response. (A) The σ32 regulon protects many cytoplasmic molecules and processes, including transcription factors. The environmental cues that regulate the transcription factors are indicated next to the curved brace. (B) The σ32 regulon also protects cytoplasmic membranes and inner membrane proteins. Note that the overexpression of many inner membrane proteins also induces the σ32 response.
Global analysis of the regulon indicated that it encodes many transcription factors in addition to the RNA polymerase binding proteins that we have just enumerated. In fact, about 12.5% of the regulon is devoted to transcription factors, and the percentage goes up to 15% if one considers Mfd and TopA, which bind to RNA polymerase and modify its transcription properties, and FimB, which inverts a DNA segment to modify the rate of switching to the on or off transcriptional state for fimbrial synthesis. This proportion is slightly higher than the fraction of the σS regulon devoted to transcription factors (8%); previously, σS was the only alternative σ factor in E. coli thought to control many transcription factors (64). The σ32 regulon contains several two-component systems and a number of transcriptional repressors, which together extend the reach of the regulon, sometimes in subtle ways. As examples, the two-component regulator PhoP/Q and the repressor TreR are transcribed by σ70 as well as by σ32 (28). Additionally, TreR is activated by PhoP/Q. PhoP/Q senses low Mg2+ and is induced by acid stress; dual control by σ's permits cells to have a greater response to acid stress at high temperature than at low temperature, possibly because combinations of stresses are more deleterious than single stresses. Likewise, the increased synthesis of TreR at high temperature by σ32 and by σ70 activated by PhoP/Q acts synergistically with σS to increase the accumulation of trehalose, which exerts both osmoprotective and heat protective effects on proteins and membranes. Trehalose synthesis is under σS control and goes through a trehalose phosphate intermediate. The σ32-transcribed and PhoP/Q-activated TreR represses the pathway for degrading external trehalose, which is imported to the cell as trehalose phosphate (22). Increased TreR synthesis presumably ensures that all trehalose phosphate is shunted toward the production of trehalose.
Perhaps the most surprising realization concerning the role of the σ32 regulon was that many of its members (∼25%) are either membrane localized or involved in membrane-relevant functions. Some relevant functions encoded by the regulon are carbonic anhydrase, necessary to provide the bicarbonate consumed in making fatty acids; components of the system for making disulfide bonds; enzymes involved in lipoprotein maturation; and membrane-localized histidine kinase sensors, transporters, and proteases. This suggests that a major function of the response is to maintain the integrity of the inner membrane upon heat stress (Fig. 5B). Interestingly, a global analysis of overproduced proteins that induce the HSR indicated that a disproportionate fraction of the inducers were membrane proteins (44). In this regard, it is intriguing that the housekeeping function of FtsH is the degradation of unassembled or damaged inner membrane proteins (24). Thus, the FtsH protease is responsible both for quality control of the membrane and for the degradation of σ32. It remains to be determined whether the fluxes through each pathway allow the two processes to be coordinated.
The HSF-mediated HSR in eukaryotes, which is similar to the σ32-mediated response in prokaryotes, has also been analyzed on the global level (18). Here too, many additional functions not related to protein homeostasis are encoded by the regulon. Interestingly, all regulatory loops currently known to control the HSR in both prokaryotes and eukaryotes are related to protein homeostasis. Two interpretations are possible. First, protein homeostasis may be the central function of the response. However, when protein homeostasis is upset, other cellular loops are likely to be perturbed as well. Therefore, the response has evolved to encompass these functions in addition to those involved in protein homeostasis. Alternatively, the response may be sensitive to numerous inputs, but only the regulatory responses related to protein homeostasis have been identified to date. In either case, the response is considerably broader than previously imagined.
EXTENDING THE LESSONS LEARNED FROM E. COLI TO OTHER ORGANISMS AND SYSTEMS
The lessons learned from the E. coli HSR can be applied to our understanding of heat shock and other stress responses in other systems. The HSR is universal, and some hsp's are among the most highly conserved proteins in the cell; however, σ32 homologues are present only in alpha-, beta-, and gammaproteobacteria (41). While heat shock transcription factors are not as highly conserved, the regulation of the HSR appears similar in most organisms. The three most widespread HSR transcription factors are σ32, HrcA, and HSF.
HSF is the eukaryotic heat shock transcription factor. HSF regulation is similar to σ32 regulation in many ways. HSF is a transcription factor that positively regulates the HSR. HSF is also directly negatively regulated by multiple chaperones, particularly Hsp70 (DnaK) and Hsp90 (reviewed in reference 43). As for E. coli, a sequestration model has been proposed to explain the mechanism of chaperone regulation of the transcription factor. However, as this model may not be sufficient for the regulation of σ32 in the simple E. coli system, it may be unlikely that this mechanism applies in the more complicated eukaryotic system. In addition to chaperone regulation, HSF can be regulated at many other levels, including but not limited to oligomerization, phosphorylation, and localization. However, due to the complexities of these regulations, it is often unclear how these components contribute to HSF regulation during stress.
The regulation of HrcA, a heat shock transcription factor that is widespread in bacteria, also has many similarities to σ32, even though it functions as a transcriptional repressor. As is the case for the σ32-mediated HSR, chaperones negatively regulate heat shock gene transcription in HrcA-dependent systems (34), albeit by a more complex route. Here, chaperones positively regulate HrcA because they are required for the proper folding of the protein. In turn, HrcA negatively represses heat shock gene transcription (reviewed in reference 42). However, it is not clear whether this system has a regulatory loop that directly responds to temperature that is comparable to what is seen for the translational regulation of σ32. One interesting unexplored possibility is that the folding of HrcA is thermosensitive; in this case, HrcA itself might be a thermosensor. As temperature-dependent effects are present in other HSRs, it is likely that there are such effects in this system as well.
While much work has been done, further analysis of the HSR is critical. We have made good progress in understanding some of the main regulators of the HSR, yet there are still as-yet-unknown components. Additionally, as these responses are universal and some components are highly conserved, HSRs provide an ideal situation, since analysis of the response in simple organisms is likely to be directly applicable to our understanding of the HSRs in general. HSRs are important for physiological processes such as cell signaling, a wide variety of pathological conditions, and evolution; therefore, expanding our understanding of the regulation of these responses is vital.
Acknowledgments
This work was supported by National Institutes of Health grant GM36278 (to C.A.G.) and an NSF graduate research fellowship (to E.G.).
REFERENCES
- 1.Arsene, F., T. Tomoyasu, A. Mogk, C. Schirra, A. Schulze-Specking, and B. Bukau. 1999. Role of region C in regulation of the heat shock gene-specific sigma factor of Escherichia coli, σ32. J. Bacteriol. 1813552-3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertani, D., A. B. Oppenheim, and F. Narberhaus. 2001. An internal region of the RpoH heat shock transcription factor is critical for rapid degradation by the FtsH protease. FEBS Lett. 49317-20. [DOI] [PubMed] [Google Scholar]
- 3.Blaszczak, A., C. Georgopoulos, and K. Liberek. 1999. On the mechanism of FtsH-dependent degradation of the sigma 32 transcriptional regulator of Escherichia coli and the role of the Dnak chaperone machine. Mol. Microbiol. 31157-166. [DOI] [PubMed] [Google Scholar]
- 4.Bugl, H., E. B. Fauman, B. L. Staker, F. Zheng, S. R. Kushner, M. A. Saper, J. C. Bardwell, and U. Jakob. 2000. RNA methylation under heat shock control. Mol. Cell 6349-360. [DOI] [PubMed] [Google Scholar]
- 5.Chae, C., S. Sharma, J. R. Hoskins, and S. Wickner. 2004. CbpA, a DnaJ homolog, is a DnaK co-chaperone, and its activity is modulated by CbpM. J. Biol. Chem. 27933147-33153. [DOI] [PubMed] [Google Scholar]
- 6.Cheng, B., C. X. Zhu, C. Ji, A. Ahumada, and Y. C. Tse-Dinh. 2003. Direct interaction between Escherichia coli RNA polymerase and the zinc ribbon domains of DNA topoisomerase I. J. Biol. Chem. 27830705-30710. [DOI] [PubMed] [Google Scholar]
- 7.Dougan, D. A., A. Mogk, K. Zeth, K. Turgay, and B. Bukau. 2002. AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 5296-10. [DOI] [PubMed] [Google Scholar]
- 8.El-Samad, H., and M. Khammash. 2006. Regulated degradation is a mechanism for suppressing stochastic fluctuations in gene regulatory networks. Biophys. J. 903749-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Samad, H., H. Kurata, J. C. Doyle, C. A. Gross, and M. Khammash. 2005. Surviving heat shock: control strategies for robustness and performance. Proc. Natl. Acad. Sci. USA 1022736-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamer, J., H. Bujard, and B. Bukau. 1992. Physical interaction between heat shock proteins DnaK, DnaJ, and GrpE and the bacterial heat shock transcription factor sigma 32. Cell 69833-842. [DOI] [PubMed] [Google Scholar]
- 11.Gamer, J., G. Multhaup, T. Tomoyasu, J. S. McCarty, S. Rudiger, H. J. Schonfeld, C. Schirra, H. Bujard, and B. Bukau. 1996. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 15607-617. [PMC free article] [PubMed] [Google Scholar]
- 12.Genevaux, P., C. Georgopoulos, and W. L. Kelley. 2007. The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol. Microbiol. 66840-857. [DOI] [PubMed] [Google Scholar]
- 13.Georgopoulos, C. 2006. Toothpicks, serendipity and the emergence of the Escherichia coli DnaK (Hsp70) and GroEL (Hsp60) chaperone machines. Genetics 1741699-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossman, A. D., D. B. Straus, W. A. Walter, and C. A. Gross. 1987. Sigma 32 synthesis can regulate the synthesis of heat shock proteins in Escherichia coli. Genes Dev. 1179-184. [DOI] [PubMed] [Google Scholar]
- 15.Gruber, T. M., and C. A. Gross. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57441-466. [DOI] [PubMed] [Google Scholar]
- 16.Guisbert, E., C. Herman, C. Z. Lu, and C. A. Gross. 2004. A chaperone network controls the heat shock response in E. coli. Genes Dev. 182812-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guisbert, E., V. A. Rhodius, N. Ahuja, E. Witkin, and C. A. Gross. 2007. Hfq modulates the σE-mediated envelope stress response and the σ32-mediated cytoplasmic stress response in Escherichia coli. J. Bacteriol. 1891963-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn, J. S., Z. Hu, D. J. Thiele, and V. R. Iyer. 2004. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 245249-5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herman, C., S. Prakash, C. Z. Lu, A. Matouschek, and C. A. Gross. 2003. Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol. Cell 11659-669. [DOI] [PubMed] [Google Scholar]
- 20.Herman, C., D. Thevenet, R. D'Ari, and P. Bouloc. 1995. Degradation of sigma 32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc. Natl. Acad. Sci. USA 923516-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horikoshi, M., T. Yura, S. Tsuchimoto, Y. Fukumori, and M. Kanemori. 2004. Conserved region 2.1 of Escherichia coli heat shock transcription factor σ32 is required for modulating both metabolic stability and transcriptional activity. J. Bacteriol. 1867474-7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horlacher, R., and W. Boos. 1997. Characterization of TreR, the major regulator of the Escherichia coli trehalose system. J. Biol. Chem. 27213026-13032. [DOI] [PubMed] [Google Scholar]
- 23.Imlay, J. A. 2006. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 591073-1082. [DOI] [PubMed] [Google Scholar]
- 24.Ito, K., and Y. Akiyama. 2005. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu. Rev. Microbiol. 59211-231. [DOI] [PubMed] [Google Scholar]
- 25.Joo, D. M., A. Nolte, R. Calendar, Y. N. Zhou, and D. J. Jin. 1998. Multiple regions on the Escherichia coli heat shock transcription factor σ32 determine core RNA polymerase binding specificity. J. Bacteriol. 1801095-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanemori, M., K. Nishihara, H. Yanagi, and T. Yura. 1997. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of σ32 and abnormal proteins in Escherichia coli. J. Bacteriol. 1797219-7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanemori, M., H. Yanagi, and T. Yura. 1999. Marked instability of the sigma(32) heat shock transcription factor at high temperature. Implications for heat shock regulation. J. Biol. Chem. 27422002-22007. [DOI] [PubMed] [Google Scholar]
- 28.Keseler, I. M., J. Collado-Vides, S. Gama-Castro, J. Ingraham, S. Paley, I. T. Paulsen, M. Peralta-Gil, and P. D. Karp. 2005. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 33D334-D337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korber, P., J. M. Stahl, K. H. Nierhaus, and J. C. Bardwell. 2000. Hsp15: a ribosome-associated heat shock protein. EMBO J. 19741-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korber, P., T. Zander, D. Herschlag, and J. C. Bardwell. 1999. A new heat shock protein that binds nucleic acids. J. Biol. Chem. 274249-256. [DOI] [PubMed] [Google Scholar]
- 31.Kurata, H., H. El-Samad, R. Iwasaki, H. Ohtake, J. C. Doyle, I. Grigorova, C. A. Gross, and M. Khammash. 2006. Module-based analysis of robustness tradeoffs in the heat shock response system. PLoS Comput. Biol. 2e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liberek, K., T. P. Galitski, M. Zylicz, and C. Georgopoulos. 1992. The DnaK chaperone modulates the heat shock response of Escherichia coli by binding to the sigma 32 transcription factor. Proc. Natl. Acad. Sci. USA 893516-3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarty, J. S., S. Rudiger, H. J. Schonfeld, J. Schneider-Mergener, K. Nakahigashi, T. Yura, and B. Bukau. 1996. Regulatory region C of the E. coli heat shock transcription factor, sigma32, constitutes a DnaK binding site and is conserved among eubacteria. J. Mol. Biol. 256829-837. [DOI] [PubMed] [Google Scholar]
- 34.Mogk, A., A. Volker, S. Engelmann, M. Hecker, W. Schumann, and U. Volker. 1998. Nonnative proteins induce expression of the Bacillus subtilis CIRCE regulon. J. Bacteriol. 1802895-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morita, M., M. Kanemori, H. Yanagi, and T. Yura. 1999. Heat-induced synthesis of σ32 in Escherichia coli: structural and functional dissection of rpoH mRNA secondary structure. J. Bacteriol. 181401-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morita, M. T., M. Kanemori, H. Yanagi, and T. Yura. 2000. Dynamic interplay between antagonistic pathways controlling the sigma 32 level in Escherichia coli. Proc. Natl. Acad. Sci. USA 975860-5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morita, M. T., Y. Tanaka, T. S. Kodama, Y. Kyogoku, H. Yanagi, and T. Yura. 1999. Translational induction of heat shock transcription factor sigma32: evidence for a built-in RNA thermosensor. Genes Dev. 13655-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagai, H., H. Yuzawa, M. Kanemori, and T. Yura. 1994. A distinct segment of the sigma 32 polypeptide is involved in DnaK-mediated negative control of the heat shock response in Escherichia coli. Proc. Natl. Acad. Sci. USA 9110280-10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagai, H., H. Yuzawa, and T. Yura. 1991. Interplay of two cis-acting mRNA regions in translational control of sigma 32 synthesis during the heat shock response of Escherichia coli. Proc. Natl. Acad. Sci. USA 8810515-10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagai, H., H. Yuzawa, and T. Yura. 1991. Regulation of the heat shock response in E coli: involvement of positive and negative cis-acting elements in translation control of sigma 32 synthesis. Biochimie 731473-1479. [DOI] [PubMed] [Google Scholar]
- 41.Nakahigashi, K., H. Yanagi, and T. Yura. 1995. Isolation and sequence analysis of rpoH genes encoding sigma 32 homologs from gram negative bacteria: conserved mRNA and protein segments for heat shock regulation. Nucleic Acids Res. 234383-4390. [PMC free article] [PubMed] [Google Scholar]
- 42.Narberhaus, F. 1999. Negative regulation of bacterial heat shock genes. Mol. Microbiol. 311-8. [DOI] [PubMed] [Google Scholar]
- 43.Nollen, E. A., and R. I. Morimoto. 2002. Chaperoning signaling pathways: molecular chaperones as stress-sensing “heat shock” proteins. J. Cell Sci. 1152809-2816. [DOI] [PubMed] [Google Scholar]
- 44.Nonaka, G., M. Blankschien, C. Herman, C. A. Gross, and V. A. Rhodius. 2006. Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev. 201776-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obrist, M., and F. Narberhaus. 2005. Identification of a turnover element in region 2.1 of Escherichia coli σ32 by a bacterial one-hybrid approach. J. Bacteriol. 1873807-3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riley, M., T. Abe, M. B. Arnaud, M. K. Berlyn, F. R. Blattner, R. R. Chaudhuri, J. D. Glasner, T. Horiuchi, I. M. Keseler, T. Kosuge, H. Mori, N. T. Perna, G. Plunkett III, K. E. Rudd, M. H. Serres, G. H. Thomas, N. R. Thomson, D. Wishart, and B. L. Wanner. 2006. Escherichia coli K-12: a cooperatively developed annotation snapshot-2005. Nucleic Acids Res. 341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rist, W., T. J. Jorgensen, P. Roepstorff, B. Bukau, and M. P. Mayer. 2003. Mapping temperature-induced conformational changes in the Escherichia coli heat shock transcription factor sigma 32 by amide hydrogen exchange. J. Biol. Chem. 27851415-51421. [DOI] [PubMed] [Google Scholar]
- 48.Selby, C. P., and A. Sancar. 1993. Molecular mechanism of transcription-repair coupling. Science 26053-58. [DOI] [PubMed] [Google Scholar]
- 49.Srivastava, R., M. S. Peterson, and W. E. Bentley. 2001. Stochastic kinetic analysis of the Escherichia coli stress circuit using sigma(32)-targeted antisense. Biotechnol. Bioeng. 75120-129. [DOI] [PubMed] [Google Scholar]
- 50.Staker, B. L., P. Korber, J. C. Bardwell, and M. A. Saper. 2000. Structure of Hsp15 reveals a novel RNA-binding motif. EMBO J. 19749-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straus, D., W. Walter, and C. A. Gross. 1990. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev. 42202-2209. [DOI] [PubMed] [Google Scholar]
- 52.Straus, D. B., W. A. Walter, and C. A. Gross. 1989. The activity of sigma 32 is reduced under conditions of excess heat shock protein production in Escherichia coli. Genes Dev. 32003-2010. [DOI] [PubMed] [Google Scholar]
- 53.Straus, D. B., W. A. Walter, and C. A. Gross. 1987. The heat shock response of E. coli is regulated by changes in the concentration of sigma 32. Nature 329348-351. [DOI] [PubMed] [Google Scholar]
- 54.Tatsuta, T., D. M. Joob, R. Calendar, Y. Akiyama, and T. Ogura. 2000. Evidence for an active role of the DnaK chaperone system in the degradation of sigma(32). FEBS Lett. 478271-275. [DOI] [PubMed] [Google Scholar]
- 55.Tatsuta, T., T. Tomoyasu, B. Bukau, M. Kitagawa, H. Mori, K. Karata, and T. Ogura. 1998. Heat shock regulation in the ftsH null mutant of Escherichia coli: dissection of stability and activity control mechanisms of sigma32 in vivo. Mol. Microbiol. 30583-593. [DOI] [PubMed] [Google Scholar]
- 56.Tomoyasu, T., F. Arsene, T. Ogura, and B. Bukau. 2001. The C terminus of σ32 is not essential for degradation by FtsH. J. Bacteriol. 1835911-5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomoyasu, T., J. Gamer, B. Bukau, M. Kanemori, H. Mori, A. J. Rutman, A. B. Oppenheim, T. Yura, K. Yamanaka, H. Niki, et al. 1995. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 142551-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomoyasu, T., T. Ogura, T. Tatsuta, and B. Bukau. 1998. Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli. Mol. Microbiol. 30567-581. [DOI] [PubMed] [Google Scholar]
- 59.Tsui, H. C., G. Feng, and M. E. Winkler. 1996. Transcription of the mutL repair, miaA tRNA modification, hfq pleiotropic regulator, and hflA region protease genes of Escherichia coli K-12 from clustered Eσ32-specific promoters during heat shock. J. Bacteriol. 1785719-5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ueguchi, C., M. Kakeda, H. Yamada, and T. Mizuno. 1994. An analogue of the DnaJ molecular chaperone in Escherichia coli. Proc. Natl. Acad. Sci. USA 911054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ueguchi, C., T. Shiozawa, M. Kakeda, H. Yamada, and T. Mizuno. 1995. A study of the double mutation of dnaJ and cbpA, whose gene products function as molecular chaperones in Escherichia coli. J. Bacteriol. 1773894-3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urech, C., S. Koby, A. B. Oppenheim, M. Munchbach, H. Hennecke, and F. Narberhaus. 2000. Differential degradation of Escherichia coli sigma32 and Bradyrhizobium japonicum RpoH factors by the FtsH protease. Eur. J. Biochem. 2674831-4839. [DOI] [PubMed] [Google Scholar]
- 63.Wade, J. T., D. C. Roa, D. C. Grainger, D. Hurd, S. J. Busby, K. Struhl, and E. Nudler. 2006. Extensive functional overlap between sigma factors in Escherichia coli. Nat. Struct. Mol. Biol. 13806-814. [DOI] [PubMed] [Google Scholar]
- 64.Weber, H., T. Polen, J. Heuveling, V. F. Wendisch, and R. Hengge. 2005. Genome-wide analysis of the general stress response network in Escherichia coli: σS-dependent genes, promoters, and sigma factor selectivity. J. Bacteriol. 1871591-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winter, J., and U. Jakob. 2004. Beyond transcription-new mechanisms for the regulation of molecular chaperones. Crit. Rev. Biochem. Mol. Biol. 39297-317. [DOI] [PubMed] [Google Scholar]
- 66.Yura, T., E. Guisbert, M. Poritz, C. Z. Lu, E. Campbell, and C. A. Gross. 2007. Analysis of {sigma}32 mutants defective in chaperone-mediated feedback control reveals unexpected complexity of the heat shock response. Proc. Natl. Acad. Sci. USA 10417638-17643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yura, T., and K. Nakahigashi. 1999. Regulation of the heat-shock response. Curr. Opin. Microbiol. 2153-158. [DOI] [PubMed] [Google Scholar]
- 68.Zhao, K., M. Liu, and R. R. Burgess. 2005. The global transcriptional response of Escherichia coli to induced sigma 32 protein involves sigma 32 regulon activation followed by inactivation and degradation of sigma 32 in vivo. J. Biol. Chem. 28017758-17768. [DOI] [PubMed] [Google Scholar]
- 69.Zhou, Y., and S. Gottesman. 1998. Regulation of proteolysis of the stationary-phase sigma factor RpoS. J. Bacteriol. 1801154-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]