

Abstract
We present a novel approach utilizing stable isotope labeling and mass spectrometric detection, which we term differential kinetic flux profiling, to probe dynamic cellular metabolic changes in response to environmental perturbations. Cells are switched into isotope-labeled media both shortly before and separately shortly after the perturbation, and the kinetics of subsequent labeling of intracellular metabolites measured by liquid chromatography-tandem mass spectrometry. The combined data enable measurement of both metabolite formation from macromolecular decomposition and small molecule flux changes triggered by the environmental perturbation. This approach is exemplified using 15N-ammonia labeling of Escherichia coli to probe changes in amino acid metabolism induced by carbon starvation. We find that carbon starvation almost fully halts amino acid fluxes, and that the trace remaining flux is almost completely driven by protein degradation. The general approach of differential kinetic flux profiling should be of wide utility for dissecting cellular metabolic responses to changing environmental conditions.
Methods for determining the route by which metabolites are generated in living cells are critical for identifying disease-associated metabolic pathways, to characterizing drug responses, and to guiding metabolic engineering efforts.1,2 Current methods can distinguish among alternative pathways of central carbon metabolism in steadily growing cells based on metabolite isotope labeling patterns following feeding of partially labeled glucose.3 They are, however, unable to adequately assess changes in metabolic fluxes that occur in response to alteration of environmental conditions. One important category of flux change, which characterizes the fed-fasting transition as well as certain drug responses, involves the main route of metabolite production shifting from de novo synthesis to macromolecule degradation.4,5 Here we present a method for quantitating this type of flux change. The general experimental approach, kinetic flux profiling (KFP), involves rapid switching of cells from unlabeled to isotope-labeled nutrient and measuring the kinetics of resulting assimilation of the label into cellular metabolites.6 The approach is applied here to study the response of the amino acid pools and fluxes of Escherichia coli to carbon starvation, a common stressor experienced by bacteria in their natural environments.7 We show that conducting KFP both shortly before and shortly after an environmental perturbation (“differential KFP”) enables for the first time reliable differentiation of metabolites formed from de novo synthesis versus macromolecule decomposition.
To enable facile manipulation of the nutrient environment of E. coli, we grow the cells on filters placed on an agarose media support. Transfer of a cell-loaded filter to media containing isotope-labeled nutrient enables non-disruptive isotope switching on a 5 s timescale.6 The ability to conduct the isotope-switch on a timescale faster than turnover of intracellular metabolite pools is a requirement for obtaining high value KFP data. The filter culture method also allows quick induction of carbon starvation by transferring the cell-loaded filter from glucose-containing minimal media to media lacking a carbon source. Within 10 min of carbon-source removal, metabolite concentration changes characteristic of carbon-starvation were triggered (e.g., ∼ 10 fold-increase for cyclic-AMP and phosphoenolpyruvate and ∼ 10 fold-decrease for fructose-bisphosphate).6,8 In contrast to the substantial alterations of the pools of glycolytic compounds early during carbon starvation, amino acid pools largely maintain homeostasis, implying balanced production (influx) and consumption (efflux) of these metabolites (Supporting Fig. 1).6
To investigate how the fluxes through amino acid pools change during carbon starvation, we performed KFP (with 15N-ammonia as the isotope-labeled nutrient) in exponentially growing E. coli (unstarved), E. coli starved of carbon for 10 min (starve-switch), and E. coli which were carbon-starved beginning 5 min after the N-isotope switch (switch-starve) (Fig. 1). For the switch-starve condition, the brief 15N-exposure prior to induction of carbon starvation labels most free amino acids without substantially labeling protein. Establishing this dichotomy (labeled amino acids, unlabeled protein) prior to the environmental perturbation proves critical to deciphering the flux changes triggered by it.
Figure 1.
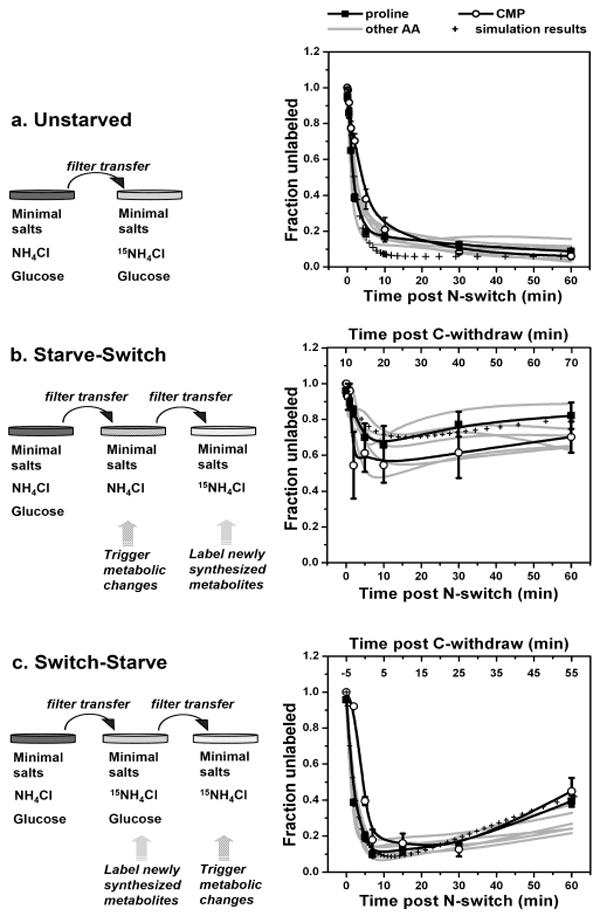
Differential-KFP of amino acid fluxes during carbon starvation of E. coli. The left column illustrates the different experimental manipulations employed and the right column the corresponding isotope labeling results. Mean data for proline and CMP are shown by symbols with error bars of ± 1 SE (n=3 independent experiments). The results for alanine, lysine, methionine, threonine, tryptophan, and valine (which were all similar to both each other and proline) are shown as the grey lines, which are a smoothed fit to the experimental data (using the B-spline function of Origin, v6.0, OriginLab Corporation, Northampton, MA). The hatched lines show the simulation of the data by the differential equation-based model provided in the Supporting Material. Fraction unlabeled = [signal of unlabeled compound]/ [signal of all forms of compound].
As shown in Figure 1a, for unstarved E. coli, incorporation of 15N into free amino acids is very fast (≥80% labeling is achieved within 10 minutes). About 8% of the free amino acids remain unlabeled even after growing on the 15N-containing media for 60 minutes, consistent with the release of unlabeled amino acids by the turnover of proteins in growing E.coli.9
If glucose (the sole carbon source) is removed from the media 10 minutes prior to the N-isotope switch, ammonia still enters the bacteria and is converted (albeit at a somewhat reduced rate compared to exponentially growing cells) into the central intermediates of N-metabolism, glutamine and glutamate (Supporting Fig. 2). Both the speed and extent of N-labeling of downstream amino acids is, however, much reduced (Fig. 1b), indicating that their de novo synthesis turns off. A trend towards a slight increase in the fraction of unlabeled compound over time (i.e., the up-swing between 10 and 60 minutes), suggests the possibility that macromolecule degradation becomes the major source of amino acids shortly after induction of carbon-starvation.
One potential interpretation of the starve-witch data (Fig. 1b) is in terms of altered amino acid production (biosynthesis and protein degradation) during carbon starvation; another is in terms of altered amino acid consumption (catabolism and/or protein synthesis). The observed approximate homeostasis of amino acid pools (Supporting Fig. 1) implies that both explanations are roughly equally correct: amino acid production and consumption change in parallel. The observed difference in labeling patterns sheds light specifically on the processes producing (but not those consuming) amino acids, however, as de novo biosynthesis versus protein degradation result in different labeling patterns. In contrast, assuming, consistent with current literature, no compartmentation of amino acids in E. coli,10 processes consuming amino acids do not discriminate between labeled and unlabeled forms.
In light of these considerations, two explanations are plausible for the results shown in Figure 1b: (1) total flux through amino acid pools essentially halts during carbon starvation (i.e., protein synthesis and degradation both happen very slowly; the observed unlabeled amino acids were produced by de novo synthesis prior to the N-isotope switch) or (2) although de novo amino acid synthesis is turned off by carbon starvation, a considerable amount of existing protein is degraded, and the resulting free amino acids are catabolized to enable energy production and consumed by proteome remodeling (the observed unlabeled amino acids were largely produced by protein degradation).
These two scenarios are distinguished by the switch-starve experiment (Fig. 1c), in which free metabolites, but not protein, were largely labeled before inducing carbon-starvation. Qualitatively, the increase in the fraction of unlabeled free amino acids upon induction of carbon starvation (despite 15N-ammonia being the sole external N-source) indicates that an increasing fraction of amino acids are generated by protein degradation.
The much higher extent of 15N-labeling at later time points of the switch-starve relative to the starve-switch experiment implies that total amino acid flux must be small since a high flux would result in rapid convergence of the fraction labeling between the two conditions. Thus, the unlabeled amino acids seen in the starve-switch condition were to a large extent members of the free amino acid pool at the time of the N-isotope switch, rather than produced by protein degradation.
Quantitatively, one can define a half-time for convergence of the metabolite labeling of cells grown under identical conditions but with different initial extents of labeling (see Supporting Information). This half-time, which is approximately proportional to pool size divided by metabolite flux, is roughly 50 min in the data obtained in carbon-starved cells and implies a decrease in total amino acid flux (de novo synthesis + protein degradation) of approximately 20-fold during carbon starvation. A simplification in the convergence half-time calculation is an assumption of fixed fluxes throughout the carbon-starvation period. An alternative quantitative treatment involves simulation of the fluxes and resulting labeling data via a system of differential equations which includes changing fluxes during the carbon-starvation period. Simulations of this sort fit the data well (Fig. 1, Supporting Fig 3), with the fit shown in Fig. 1 involving first order decay of the de novo flux to zero with a half-time of 4 min during carbon starvation and exponential decay of the degradation flux to ∼20% of its initial value with the same half-time. While a diversity of specific flux patterns can fit the data, all involve a substantial reduction of the total flux with the degradation flux decreasing less than the biosynthetic one. The detailed kinetic patterns corresponding to different amino acids in Figure 1 may also reflect compound-specific factors, such as their extent of residual biosynthesis, their abundance in the proteins being made and degraded, and the absolute magnitude of their pool size.
Similar patterns to those observed for the amino acids were also found for cytidine monophosphate (CMP) (Fig. 1 open circles), suggesting that during carbon starvation, the total flux of nucleotides into RNA likely also decreases profoundly, with a concomitant increase in the fraction of the nucleotide flux coming from RNA degradation relative to de novo synthesis. More complete nucleotide information was not obtained in the present experiment, however, due to the limited ability of cold methanol to extract nucleotides from E. coli.11 The similarity of the CMP and proline patterns likely reflects their having (by chance) similar pool sizes relative to fluxes; the precise pattern for other nucleotides is likely to be different.
The observation of decreased biosynthetic flux in response to carbon starvation was expected. The relatively slow protein degradation flux in the starved cells was less so, given prior literature12,13 reporting increased protein degradation in starved E. coli. The modest protein degradation flux measured here would, nevertheless, result in substantial proteome degradation over long starvation intervals (e.g. > 4 hours) as observed previously. Much of the protein degradation flux in growing cells likely arises from fast turnover of a small pool of very short lived proteins.9 Turnover of these proteins likely slows during carbon starvation (resulting in a decrease in protein degradation flux), while the fraction of the proteome subject to degradation likely in turn increases. One particular case of a normally short-lived protein being stabilized by starvation involves the key transcriptional regulator RPOS.14 Evolving proteomic technologies, when paired with isotopic labeling techniques such as those described here, are poised to enable determination of the lifetimes of the full spectrum of proteins in both starved and unstarved conditions in the near future.15,16 Combining proteomic and metabolomic approaches should provide a yet more complete picture of E. coli's starvation behavior.
In summary, differential-KFP takes advantage of the unique ability of isotopes to shed light on molecules that are chemically identical but have different “histories” (e.g. sources and times of synthesis) to distinguish metabolites generated by macromolecule degradation from those synthesized de novo. The same strategy can presumably be applied to different classes of macromolecules and their building blocks to gain insights about other physiological changes. For example, using isotope-labeled glucose, differential-KFP should thus be able to distinguish conditions which halt glycolytic flux from those which trigger a switch from use of environmental glucose to intracellular glycogen as the carbon source. Although demonstrated using E.coli, this approach should be applicable to a wide spectrum of cell types including mammalian cells in culture. For metabolites that exist as separated pools in distinct eukaryotic intracellular compartments, however, mechanistic interpretation of the data will need to take this into account. Despite this complication, differential kinetic flux profiling is poised to contribute to better understanding of dynamic metabolic responses to a wide diversity of environmental conditions and signals.
Supplementary Material
Response of metabolite pools to carbon starvation in E.coli, labeling kinetics of glutamine and glutamate, derivation of half-time for convergence, model construction, matlab code and output.
Acknowledgments
We thank the Beckman Foundation, the NIH Center for Quantitative Biology (P50 GM071508), the NSF DDDAS-SMRP program, and the American Heart Association for their support, and Bryson Bennett and Shawn Campagna for their helpful comments.
References
- 1.Hellerstein MK. Annu Rev Nutr. 2003;23:379–402. doi: 10.1146/annurev.nutr.23.011702.073045. [DOI] [PubMed] [Google Scholar]
- 2.Kern A, Tilley E, Hunter IS, Legisa M, Glieder A. J Biotechnol. 2007;129:6–29. doi: 10.1016/j.jbiotec.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 3.Sauer U. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.St John AC, Goldberg AL. J Bacteriol. 1980;143:1223–33. doi: 10.1128/jb.143.3.1223-1233.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. Oncogene. 2005;24:4165–73. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 6.Yuan J, Fowler WU, Kimball E, Lu W, Rabinowitz JD. Nat Chem Biol. 2006;2:529–30. doi: 10.1038/nchembio816. [DOI] [PubMed] [Google Scholar]
- 7.Koch AL. Adv Microb Physiol. 1971;6:147–217. doi: 10.1016/s0065-2911(08)60069-7. [DOI] [PubMed] [Google Scholar]
- 8.Brauer MJ, Yuan J, Bennett BD, Lu W, Kimball E, Botstein D, Rabinowitz JD. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0609508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch AL, Levy HR. J Biol Chem. 1955;217:947–57. [PubMed] [Google Scholar]
- 10.Koch AL. Annu Rev Microbiol. 1996;50:317–48. doi: 10.1146/annurev.micro.50.1.317. [DOI] [PubMed] [Google Scholar]
- 11.Kimball E, Rabinowitz JD. Anal Biochem. 2006;358:273–80. doi: 10.1016/j.ab.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nath K, Koch AL. J Biol Chem. 1971;246:6956–67. [PubMed] [Google Scholar]
- 13.Mandelstam J. Nature. 1957;179:1179–81. doi: 10.1038/1791179a0. [DOI] [PubMed] [Google Scholar]
- 14.Pratt LA, Silhavy TJ. Proc Natl Acad Sci U S A. 1996;93:2488–92. doi: 10.1073/pnas.93.6.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cargile BJ, Bundy JL, Grunden AM, Stephenson JL., Jr Anal Chem. 2004;76:86–97. doi: 10.1021/ac034841a. [DOI] [PubMed] [Google Scholar]
- 16.Beynon RJ. Brief Funct Genomic Proteomic. 2005;3:382–90. doi: 10.1093/bfgp/3.4.382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Response of metabolite pools to carbon starvation in E.coli, labeling kinetics of glutamine and glutamate, derivation of half-time for convergence, model construction, matlab code and output.