

Abstract
Tissue type transglutaminase (TG2) is a unique multifunctional protein that plays a role in many steps in the cancer metastatic cascade. Here, we examined the clinical (n = 93 epithelial ovarian cancers) and biological (in vitro adhesion, invasion, and survival and in vivo therapeutic targeting) significance of TG2 in ovarian cancer. The overexpression of TG2 was associated with significantly worse overall patient survival in both univariate and multivariate analyses. Transfection of TG2 into the SKOV3ip1 cells promoted attachment and spreading on fibronectin-coated surfaces and increased the in vitro invasive potential of these cells. Conversely, TG2 silencing with siRNA of HeyA8 cells significantly decreased the invasive potential of the cells, and also increased docetaxel-induced cell death. In vivo therapy experiments using chemotherapy-sensitive (HeyA8) and resistant (HeyA8-MDR, RMG2) models demonstrated significant anti-tumor activity both with TG2 siRNA-DOPC alone and in combination with docetaxel chemotherapy. This anti-tumor activity was related to decreased proliferation, angiogenesis and increased tumor cell apoptosis in vivo. Taken together, these findings indicate that TG2 overexpression is an adverse prognostic factor in ovarian carcinoma and TG2 targeting may be an attractive therapeutic approach.
Introduction
Based on current cancer statistics, ovarian cancer remains the most common cause of death from a gynecologic malignancy in the United States (1). This poor outcome relates to the fact that most patients present with advanced stage disease and widespread peritoneal metastasis. While most ovarian cancer patients respond to initial therapy of cytoreductive surgery and platinum-based chemotherapy, more than 70% will recur and eventually succumb to disease (2). Therefore, understanding the molecular factors responsible for ovarian cancer metastasis and development of novel therapeutic approaches is urgently needed.
Tissue transglutaminase (TG2) is a unique multifunctional protein that can catalyze Ca2+-independent hydrolysis of guanosine triphosphate(GTP) and adenosine triphosphate, the protein disulfide isomerase reaction (3), and Ca2+-dependent post-translational modification of proteins (4). TG2 also has serine/threonine kinase activity (5–7). The ability of TG2 to hydrolyze GTP enables it to serve as a signaling molecule, leading to the activation of a cytoplasmic target, phospholipase C (8). Although mainly a cytoplasmic protein, TG2 can also be secreted outside the cell where it regulates cell–matrix interactions (9) and can translocate to the nucleus where it associates with pRb, p53 and histones to regulate certain cellular functions (10–12). Importantly, TG2 can also exist on the cell surface membrane in association with specific integrin family members (beta1, beta3, beta4, and beta5), for which TG2 serves as a coreceptor, and promotes stable interactions between cells and the extracellular matrix (ECM), resulting in increased integrin-mediated cell adhesion, spreading, migration and survival functions (13, 14). TG2 has been shown to activate RhoA and mitogen-activated protein kinase pathways (MAPK), which control key downstream signaling steps that affect the invasive and metastatic behavior of malignant cells (15). TG2 overexpression has been noted in several cancers including malignant melanoma (16), breast cancer (17, 18), and pancreatic cancer (19). In these cancers, TG2 contributes to the development of chemoresistance by exploiting integrin-mediated cell survival signaling pathways. In addition TG2 overexpression contributes to cancer cell adhesion and invasion. Based on these features, TG2 appears to be an attractive therapeutic target.
We have recently developed a highly efficient in vivo method for systemic delivery of small interfering RNA (siRNA) by using a neutral liposome, 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC; ref. 20). Our proof-of-concept studies with targeting EphA2 (20) or FAK (21) demonstrated the therapeutic efficacy of this approach. In the current manuscript, we demonstrate the clinical significance of TG2 as a prognostic factor for ovarian cancer patients. In addition, we characterize the biological effects of TG2 on ovarian cancer progression and utilize a therapeutically relevant approach for TG2 silencing.
Materials and Methods
Ovarian cell lines and culture condition
The ovarian cancer cell lines HeyA8, SKOV3ip1 (20, 21), A2780-PAR, A2780-CP20, OVCAR3, IGROV1, ES2, 222, and RMG2 were cultured in RPMI 1640 supplemented with 10–15% fetal bovine serum and 0.1% gentamicin sulfate (Gemini Bioproducts, Calabasas, CA). The taxane-resistant HeyA8-MDR (a kind gift from Dr. Isaiah Fidler, Department of Cancer Biology, University of Texas M. D. Anderson Cancer Center) and SKOV3-TR cells were maintained in paclitaxel (300 ng/mL for HeyA8-MDR, 150 ng/mL for SKOV3-TR) containing media. The non-transformed ovarian surface epithelial cell line, HIO-180 (22), was a kind gift from Dr. Andrew Godwin (Fox Chase Cancer Center, Philadelphia, PA). All in vitro experiments were conducted with 60 to 80% confluent cultures. The therapy experiments were performed with 3 ovarian cancer cell lines: HeyA8, HeyA8-MDR, and RMG2. For in vivo injections, the cells were washed twice with PBS, detached with 0.1 % EDTA, centrifuged at 1,100 rpm for 7 minutes at 4°C, and reconstituted in serum-free HBSS (Life Technologies, Carlsbad, CA) at a concentration of 1.25 × 106 cells/mL (HeyA8) or 5.0 × 106 cells/mL (HeyA8-MDR) or 17.5 × 106 cells/mL (RMG2) for 200 μL intraperitoneal (i.p.) injection volume per one mouse. Only single-cell suspensions with more than 95% viability, as determined by trypan blue exclusion, were used for the in vivo injections.
Drugs and reagents
Leupeptin, aprotinin, and sodium orthovanadate were obtained from Sigma (St. Louis, MO), EDTA was from Life Technologies/Invitrogen (Carlsbad, CA). Paclitaxel was from Bristol-Myers Squibb (Princeton, NJ) and docetaxel was from Sanofi-Aventis (Bridgewater, NJ). Primary antibodies used were mouse anti-TG2 monoclonal CUB-7401 (Neomarkers, Fremont, CA), mouse anti–proliferating cell nuclear antigen (PCNA) clone PC 10 (DAKO A/S, Copenhagen, Denmark), and mouse anti-CD31 (PharMingen, San Diego, CA). Rabbit polyclonal antibodies to phosphorylated Akt (pAkt; Ser473) and Akt were from Cell Signaling Technology (Beverly, MA). The following secondary antibodies were used for colorimetric immunohistochemical analysis: Horse radish peroxidase (HRP)-conjugated goat anti-rabbit IgG F(ab′)2 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), biotinylated mouse anti-goat (Biocare Medical, Walnut Creek, CA), HRP-conjugated streptavidin (DAKO), HRP-conjugated rat anti-mouse IgG2a (Serotec, Harlan Bioproducts for Science, Inc., Indianapolis, IN), HRP-conjugated goat anti-rat IgG (Jackson ImmunoResearch Laboratories), and fluorescent Alexa 488–conjugated goat anti-rabbit IgG (Molecular Probes, Inc., Eugene, OR).
Western blot and TG2 enzymatic activity
Cell lysates were made using modified radioimmunoprecipitation assay (RIPA) buffer (compositions; 50 mM/L Tris, 150 mM/L NaCl, 1% Triton, 0.5% deoxycholate, 25 μg/mL leupeptin, 10 μg/mL aprotinin, 2 mM/L EDTA, and 1 mM/L sodium orthovanadate) as described previously (22, 23). To prepare lysates of snap-frozen tissue from mice, 30 mm3 cuts of tissue were disrupted using tissue homogenizer, and centrifuged at 13,000 rpm for 30 minutes. The total protein concentration of the supernatant was determined using a bicinchoninic acid protein assay reagent kit (Pierce, Rockford, IL). Proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membrane by wet transfer system (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked with 5% non-fat milk and incubated with anti-TG2 antibody over night at 4°C. HRP-conjugated anti-mouse IgG (Amersham, Piscataway, NJ) was applied, and developed with enhanced chemiluminescence detection kit (Pierce Biotechnology). The anti-β-actin primary antibody (Sigma) was used to confirm equal loading. For analyzing TG2 enzymatic activity, cells were harvested in lysis buffer (20 mM/L Tris-HCl with pH 7.4 containing 1 mM/L EDTA, 150 mM/L NaCl, 14 mM/L 2-mercaptoethanol, 1mM L-phenylmethylsulfonyl fluoride) by probe sonification. TG2 activity was assayed by determining the calcium-dependent incorporation of [H3] putrescine (specific activity, 14.3 Ci/mmol; Amersham Pharmacia, San Fransisco, CA) into dimethylcasein as described previously (24). The enzyme activity was expressed as nanomoles of putrescine incorporated per milligram of total cell protein.
TG2 Immunohistochemical staining of clinical samples and mouse tissue with statistical analyses
The 93 ovarian cancer samples from M. D. Anderson gynecologic oncology tumor bank were used for the clinical relevance. Sections of formalin-fixed, paraffin-embedded tumor samples (5 μm thick) were heated to 60°C and dehydrated in xylene and graded alcohols. Antigen retrieval was performed with 0.1 M citrate buffer at pH 6.0 for 30 minutes in a 95°C steamer. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide. To prevent non-specific binding to Fc receptor of the mouse tissue by the primary antibody made in mouse, mouse IgG Fc blocker (The Jackson Laboratory, Bar Harbor, ME) was applied, followed by incubation with anti-TG2 monoclonal antibodies, CUB-7401. After incubation for 30 minutes each with biotinylated secondary antibody and peroxidase-labeled streptavidin, antigen-antibody reactions were detected by exposure to 3,3′-diaminobenzidine (DAB; Phoenix Biotechnologies, Huntsville, AL) for 3–5 minutes. The immunostained slides were examined under the light microscope and scored independently by two researchers and one pathologist. The scoring was done using staining intensity (low, 1; moderate, 2; high, 3) and the percentage of positive cancer cells (≤ 5 %, 0; 6–25 %, 1; 26–50 %, 2; 51–75 %, 3; ≥ 76 %, 4). A composite score was generated as the sum of these two parameters and the expression was dichotomized as: TG2 overexpression (overall score > 4) or low/absent expression (overall score ≤ 4). The 2 test was used to determine differences among variables using of the Statistical Package for the Social Sciences (SPSS; SPSS Inc., Chicago, IL). Kaplan–Meier survival plots were generated, and comparisons between survival curves were made with the log-rank statistic. The Cox proportional hazards model was used for multivariate analysis. A p value < 0.05 was considered statistically significant.
Wild-type and C277S mutant TG2 adenovirus generation
An adenovirus containing full-length TG2 (TG2-W) or C277S mutant (TG2-M) cDNA was kindly provided by Dr. Ugra Singh (The Texas A&M University System Health Science Center, Temple, TX). TG2 cDNA cloned in pcDNA3.1 vector was first subcloned in a pshuttle 2 vector and then in a BD adenoX adenoviral vector. Human embryonic kidney (HEK293) cells were transfected with recombinant adenoviral plasmid for packaging of adenovirus particles followed by purification of adenovirus on CsCl gradient and used at 25 multiplicities of infection (MOI). Cells which were infected with lacZ adenovirus served as control. The SKOV3ip1 cells, which had low baseline TG2 expression, were infected with adenovirus alone (EV) or TG2-W or TG2-M. After 48h, TG2 activity and protein expression were determined. Same cells were used for cell attachment, invasion, and survival assays. The SKOV3ip1 cells were selected for these experiments because from our experience, we have found that cells with complete lack of TG2 expression are not able to sustain TG2 expression (presumably due to low GTP and/or perturbation of cytosolic free calcium resulting from transfection, leading to cross-linking of proteins and cell death).
Cell attachment, invasion and survival
For adhesion assays, cells (3 × 104 cells/well/0.2 mL serum-free RPMI-1640 medium) were incubated in fibronectin (Fn)- or bovine serum albumin (BSA)-coated 96-well plates. After an hour of incubation at 37 °C, non-adherent cells were removed by washing with PBS and attached cells were stained with Crystal violet, observed under a microscope for morphologic analysis and optical density was measured at 540 nm for quantitative analysis. The invasive potential of cells was determined in vitro by using Matrigel-transwell inserts as described earlier (25). Briefly, Matrigel-transwell inserts with a 12-μm pore size were coated with 0.78 mg/mL Matrigel in cold serum-free RPMI-1640 medium. The cell suspension (1 × 106 cells) was added to duplicate transwells. After incubation for 48 hours, the cells that passed through the filter on the underside of the membrane were stained and counted under a light microscope. Ten fields of cells were counted for each well, and the mean number of cells per field was calculated. The number of viable cells remained after appropriate treatment was determined by measuring their ability to reduce 3-(4-5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTT) into a soluble formazan. The cells were plated on Fn-coated 96 well plates. After 12–24 hours of incubation of cells for attachment, the medium was exchanged with increasing concentrations of drugs (0 to 25 nM of paclitaxel or 0 to 50 nM of docetaxel). After incubation for 72 hours in the presence of drugs, cells were incubated with 0.15% MTT for 2 hour at 37 °C. The supernatant was removed, cells were dissolved in 100 μL DMSO and the optical absorbance was recorded at 570 nm. Each experiment was repeated thrice in triplicate.
TG2 down-regulation by siRNA and liposomal siRNA preparation
TG2-targeted specific siRNA sequence (5′-AAGGGCGAACCACCTGAACAA-3′) was synthesized and then purified using high-performance liquid chromatography by Qiagen-Xeragon (Germantown, MD). A sequence (5′-AATTCTCCGAACGTGTCACGT-3′) that did not have homology to any human mRNA as determined by blast search served as a control compared to a TG2 targeted sequence. At 70% confluence, HeyA8 cells were transfected with either control siRNA or TG2 specific siRNA as described earlier (17). Briefly, 3 × 105 cells were plated in each well of six-well plates and allowed to adhere for 24 hours. On the day of transfection, cells were washed, transfected and harvested at different time intervals. The down regulation of TG2 gene and protein expression was determined by using both RT-PCR and Western blot. For in vivo delivery, siRNA was incorporated into the phospholipid DOPC as described previously (20). Before in vivo administration, this preparation was hydrated with normal (0.9 %) saline at a concentration of 15 μg/mL to achieve the desired dose in 150 to 200 μL per injection.
Generation of orthotopic in vivo model and tissue collection after therapy
Female athymic nude mice (NCr-nu) were purchased from the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed and maintained under specific pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the U.S. Department of Agriculture, U.S. Department of Health and Human Services, and NIH. All studies were approved and supervised by the University of Texas M. D. Anderson Cancer Center Institutional Animal Care and Use Committee. The mice used in these experiments were 8 to 12 weeks old.
To evaluate the therapeutic effect of combination TG2 siRNA and docetaxel in our mouse model, we first performed preliminary dose-finding experiments for TG2 siRNA. HeyA8 (0.5 X 106) cells were injected i.p. and treatment was initiated 18 days following tumor cell injection when tumors could be assessed by palpation. Mice (n = 3 per group) were given two injections of TG2 siRNA-DOPC 3.5 μg (105 μg/kg) or 5 μg (150 μg /kg) and the tumor was harvested at various time intervals. The i.p. route for siRNA delivery was selected based on comparable uptake and therapeutic efficacy with either i.p. or i.v. routes in 200 μL volume (26). For therapy experiments, docetaxel was injected once weekly at 50 μg per mouse based on previous experiments for docetaxel kinetics for HeyA8 cells (21) and similar IC50 levels of docetaxel in both HeyA8 and RMG2 cells (data not shown). Following treatment, the mice were sacrificed at either 48 hours, 96 hours or 6 days. Mouse weight, tumor weight, number of nodules, and tumor site were recorded. Tissue specimens were snap frozen for lysate preparation, fixed in formalin for paraffin embedding, or frozen in OCT compound (Miles, Inc., Elkhart, IN) for frozen slide preparation. Immunohistochemical and Western blot analysis were done on tissue specimens to determine adequate dosage of TG2 siRNA-DOPC for further in vivo therapy experiments.
Based on the results of our preliminary dose-response experiments, we initiated a series of separate therapy experiments using the optimal TG2 siRNA dosage (50 mice per each cell line and 10 mice per each treatment group were used). Tumor cells (0.25 × 106 HeyA8 or 1.0 × 106 HeyA8-MDR or 3.5 × 106 RMG2 cells/mouse) were injected i.p., and 7 days later, mice were randomly assigned to five treatment groups: empty liposome twice weekly, nonspecific control siRNA-DOPC (150 μg/kg) twice weekly, control siRNA-DOPC in combination with docetaxel, TG2 siRNA-DOPC (150 μg/kg) twice weekly, and TG2 siRNA-DOPC plus docetaxel. Mice were monitored for adverse effects, and tumors were harvested after 3 to 4 weeks of therapy. If animals in any group became moribund, then the experiment was terminated and all animals were sacrificed together. Mouse weight, tumor weight, number of nodules, and tumor site were recorded. Tissue specimens were collected as described above.
Tissue staining for PCNA, microvessel density, and apoptosis
For immunohistochemical analysis, paraffin-embedded tissues were sectioned (8 μm thick) and used to detect expression of proliferating cell nuclear antigen (PCNA). Frozen sections were used for detecting CD31 and terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL). Generally, formalin-fixed, paraffin-embedded sections were deparaffinized in xylene, treated with a graded series of alcohol. Antigen retrieval was done by microwave heating for 5 minutes at 95°C in 0.1 M citrate buffer (pH 6.0) followed by blocking of endogenous peroxide with 3% hydrogen peroxide in methanol for 5 minutes. Incubation with primary antibody (anti-PCNA, PC-10, mouse IgG) in blocking solution was done overnight at 4°C. After two PBS washes, the appropriate secondary antibody conjugated to HRP in blocking solution was added for 1 hour at room temperature. HRP was detected with DAB substrate for 5 minutes, washed, and counterstained with Gill No. 3 hematoxylin (Sigma) for 15 seconds. To analyze microvessel density in tumor tissue, immunohistochemistry for CD31 was performed on freshly cut frozen tissue. These slides were fixed in cold acetone for 10 minutes and did not require antigen retrieval. The primary antibody used was mouse anti-CD31 (platelet/endothelial cell adhesion molecule-1, rat IgG; PharMingen).
For examination of the level of apoptosis in tumor tissue, terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) staining was performed. Briefly, freshly cut frozen tissue was fixed by 4 % paraformaldehyde in PBS for 10 minutes followed by adding 0.2 % TritonX-100 for 15 minutes at room temperature. A positive control slide was treated with DNase (1:50) dilution. Slides were incubated with TdT enzyme(1 μL/slide) for 1 hour at 37°C followed by counter staining with Hoechst. Staining for PCNA, CD31, and TUNEL was conducted on tumors collected at the conclusion of 3 or 4 weeks of therapy.
Microscopic quantitative analyses of PCNA, microvessel density, and TUNEL
To quantify microvessel density (MVD), 5 random 0.159-mm2 fields at ×100 final magnification per one slide were examined for each tumor (one slide per mouse, five slides per each treatment group) and the number of microvessels per field was counted by two investigators in a blinded fashion. A single microvessel was defined as a discrete cluster or single cell stained positive for CD31 with the presence of a lumen. To quantify PCNA expression, the number of PCNA positive and total tumor cells were counted in 5 random 0.159-mm2 fields at ×100 magnification followed by calculation of the positive cell percentages. For quantification of TUNEL positive cells, the number of TUNEL-positive and total tumor cells were counted in random 0.011-mm2 fields at ×400 magnification followed by calculation of the positive cell percentages. DAB-stained sections were examined on a Microphot-FX microscope (Nikon, Garden City, NY) equipped with a three-chip charge-coupled device color video camera (model DXC990, Sony, Tokyo, Japan). Immunofluorescence microscopy was performed using a Microphot-FXA microscope (Nikon). Images were captured using a cooled charge-coupled device camera (model 5810, Hamamatsu, Bridgewater, NJ) and Optimas Image Analysis software (Media Cybernetics, Silver Spring, MD).
Statistics
For in vivo experiments, differences in continuous variables (mean body weight, tumor weight, number of nodules, MVD, PCNA, and TUNEL) were analyzed using the Student’s t-test for comparing two groups and by ANOVA for multiple group comparisons with p < 0.05 considered statistically significant. For values that were not normally distributed, the Mann-Whitney rank sum test was used. The SPSS software was used for all statistical analyses.
Results
TG2 expression in ovarian cell lines and clinical samples
We first examined TG2 expression using Western blot in multiple ovarian cell lines (Figure 1A). The non-transformed HIO-180 cells had absent TG2 expression by Western blot analysis. Moderate to high TG2 expression was noted in the HeyA8, HeyA8-MDR, RMG2, and ES2 cells. All of the remaining cell lines had either low or absent TG2 expression. Consistent with the expression data, TG2 enzyme activity was absent or low in the A2780-PAR, A2780-CP20, and SKOV3ip1 cells. Conversely, HeyA8 and HeyA8-MDR cells demonstrated high TG2 enzyme activity (Figure 1B).
Figure 1.
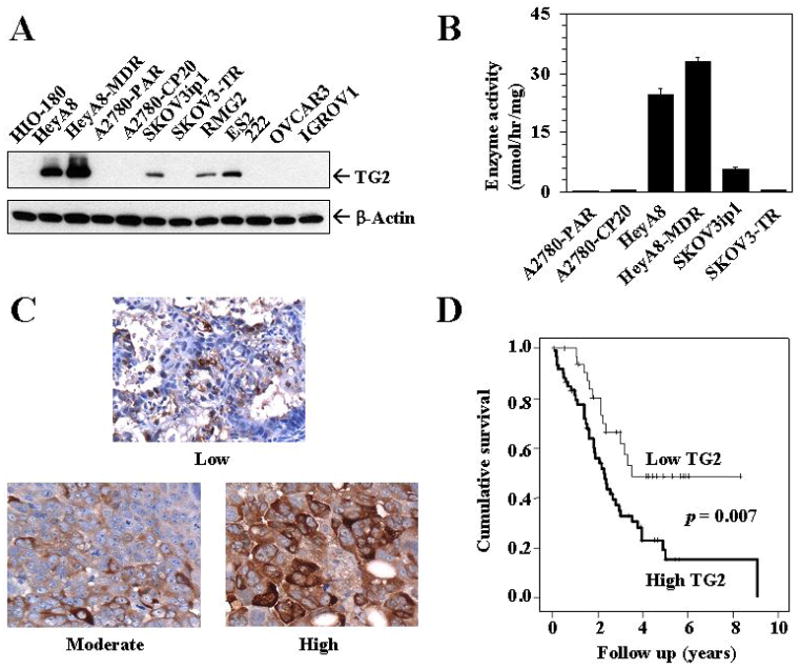
A) Western blot showing basal expression of TG2 protein in 11 ovarian cancer cell lines and one non-transformed normal ovarian epithelial cell line (HIO-180). B) Transglutaminase enzymatic activity according to TG2 expression levels. Mean enzymatic activity of TG2 was determined in the cell extract by studying Ca2+-dependent incorporation of [3H]putrescine into dimethylcasein, as described in Materials and methods. Error bars represent S.D. C) Representative immunohistochemical staining of clinical specimens for TG2 expression. All pictures were taken at original magnification × 200. D) Kaplan–Meier survival curve of patients with invasive ovarian cancer according to TG2 expression.
Based on high TG2 expression in several ovarian cancer cell lines, we next examined TG2 expression in 93 epithelial ovarian cancers using immunohistochemistry. Representative examples of TG2 staining are shown in Figure 1C and correlations with clinicopathological variables are listed in Table 1. The demographic features of the patients with invasive ovarian cancers are listed in Supplementary Table 1. TG2 overexpression was associated with high tumor stage (69% of high-stage ovarian cancers overexpressed TG2 compared with 30% of low-stage cancers; p = 0.01). TG2 expression was not related to histological subtype (serous versus other), grade (low versus high), presence of ascites, or level of cytoreduction (optimal versus suboptimal). In univariate analysis, TG2 overexpression was associated with significantly worse patient survival (median survival 2.29 years for those with high TG2 expression versus not yet reached for individuals with low expression, p=0.007; Figure 1D). In multivariate analysis including age, stage, grade, level of cytoreduction, and TG2 expression, only stage (p<0.002), level of cytoreduction (p=0.001), and TG2 overexpression (p<0.04) were independent predictors of poor survival (Supplementary Table 2).
Table 1.
Correlation of clinicopathological variables with TG2 overexpression in patients with invasive ovarian cancer
| TG2 overexpression
|
|||
|---|---|---|---|
| Variable | No | Yes | p |
| Stage | |||
| Low (I or II) | 7 | 3 | 0.01 |
| High (III or IV) | 25 | 58 | |
| Histology | |||
| Serous | 25 | 54 | 0.06 |
| Other | 8 | 6 | |
| Grade | |||
| Low | 3 | 2 | 0.48 |
| High | 29 | 59 | |
| Ascites | |||
| Yes | 24 | 49 | 0.28 |
| No | 9 | 11 | |
| Cytoreduction | |||
| Optimal (<1 cm residual) | 26 | 30 | 0.29 |
| Suboptimal | 10 | 27 | |
The roles of TG2 on adhesion, invasion and survival function of cancer cells
Based on the impact of TG2 overexpression on patient outcome, we next characterized its biological effects on cancer cells. TG2 was ectopically overexpressed in the SKOV3ip1 cells, which have low baseline levels of TG2, using wild-type TG2 or mutant-type TG2 or empty vector. The overall expression of TG2 in SKOV3ip1 cells transfected with TG2-W increased by 3–4-fold over the basal levels (Supplementary Figure 1). First, we tested whether TG2 expression in ovarian cancer cells can promote adhesion to matrix, since TG2 can serve as a co-receptor for integrin-mediated binding of cells to fibronectin (14). Infection of cells with EV had no effect on cell adhesion, whereas SKOV3ip1 cells transfected with TG2-W showed 1.7-fold increased attachment and spreading when cultured on fibronectin-coated plates (p<0.001, Figure 2A). On BSA-coated plates, however, they were far less adherent and showed a rounded morphology (data not shown). We also examined effects of TG2 on the in vitro invasive potential of the transfected SKOV3ip1 cells. TG2-W transfection was associated with a parallel increase in the invasive potential of SKOV3ip1 cells (p<0.001; Figure 2B). Next, we determined if catalytic (transamidation) activity of TG2 is essential for inducing invasiveness in SKOV3ip1 cells. Infection of cells with mutant-TG2-adenoviral construct (TG2-M) coding enzymatically inactive TG2 protein as a result of point mutation in the active-site cysteine residue (Cys277Ser), promoted similar increase in invasiveness of SKOV3ip1 cells (Figure 2B). These results suggest that TG2 expression plays an important role in promoting the invasive phenotype in ovarian cancer cells and that the transamidation activity of TG2 is not essential for conferring these functions. Infection of cells with EV, however, had no effect on the invasive potential.
Figure 2.
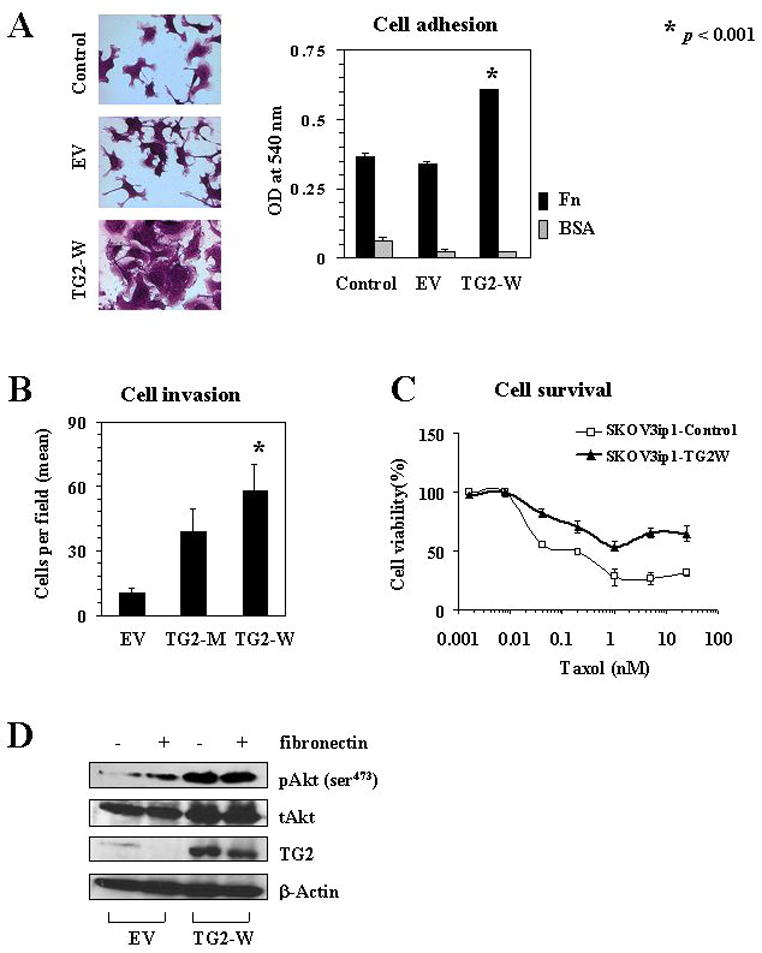
A) TG2 was ectopically introduced into SKOV3ip1 cells by adenovirus containing TG2 gene. Effects of TG2 overexpression on adhesion were examined following transfection of wild type TG2 gene (TG2-W) or empty vector (EV) into SKOV3ip1 cells. Control represents untransfected cells. Left panel pictures were obtained from fibronectin-coated plates. Right panel presents mean optical density. Error bars represent S.D. B) Effect of wild type TG2 gene (TG2-W) versus mutant type TG2 gene (TG2-M) on SKOV3ip1 invasive potential was examined using a Matrigel-transwell assay. Each experiment was performed in triplicate and repeated at least two times. C) SKOV3ip1 cells infected with TG2-W or without any treatment were plated on 96 well plates. After attachment, the medium was exchanged with increased concentrations of paclitaxel (0 to 25 nM). The number of viable cells remaining was determined after 72 hours. Error bars represent S.D. D) Western blot analysis for phosphorylated Akt (pAkt, ser473) and total Akt (tAkt) in SKOV3ip1 cells transfected with TG2-W or EV.
The interaction of cancer cells with ECM is known to induce cell survival signaling pathways and confer chemoresistance (27). Therefore, we examined whether TG2-mediated attachment of cells could confer protection from drug-induced cytotoxic effects. We found that SKOV3ip1 cells with TG2-W were more resistant to paclitaxel-induced cell death than controls (Figure 2C). Empty vector transfected cells were similar to non-transfected controls in response to paclitaxel (data not shown). These results suggest that TG2 expression in ovarian cancer cells promotes cell surface interaction with ECM and protects cells from apoptosis.
It has been suggested that TG2 expression can induce constitutive activation of focal adhesion kinase (FAK) and its downstream PI3K/Akt survival pathway, which is independent of its enzymatic activity (19). Therefore, we examined the effects of TG2-W overexpression in SKOV3ip1 cells for activation of this pathway. Western blot analysis for phosphorylated Akt (pAkt) and total Akt (tAkt) showed that overexpression of TG2 in SKOV3ip1 cells induced marked increase in pAkt (Ser473) without change in tAkt levels (Figure 2D).
Based on the effects of ectopic TG2 expression on SKOV3ip1 invasion, we next asked whether TG2 silencing would block these effects. For these experiments, we used a siRNA targeted to TG2. First, we performed in vitro TG2 knockdown experiments using HeyA8 cells with high TG2 expression. TG2 expression was decreased by 80% at 48 hours after TG2 siRNA transfection (Figure 3A and Supplementary Figure 2). HeyA8 cells were highly invasive without transfection, however, the inhibition of TG2 by siRNA significantly decreased the ability of cells to invade through the Matrigel-coated transwell inserts (p<0.001, Figure 3B), suggesting that TG2 expression promotes invasive functions in ovarian cancer cells. Also, knockdown of TG2 with siRNA increased docetaxel-induced cell death when the cells were cultured on fibronectin-coated plates (Figure 3C). These results suggest that the TG2-dependent interaction between ovarian cancer cells and fibronectin is critical for inducing cell growth and survival.
Figure 3.
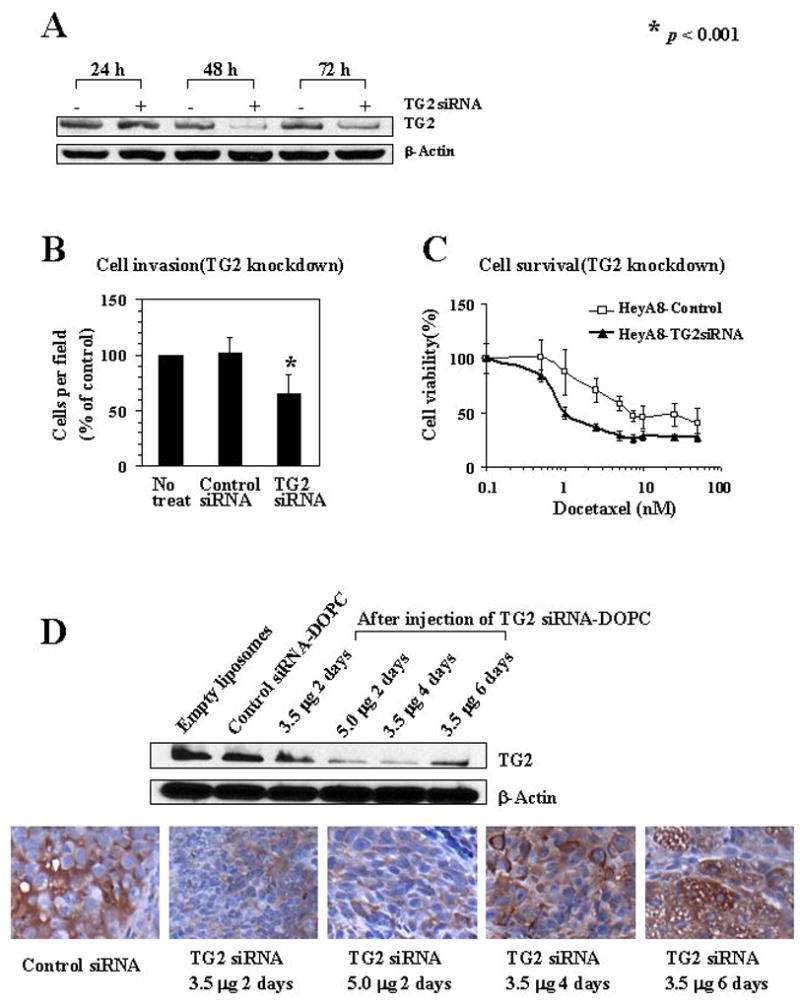
A) In vitro knockdown of TG2 in HeyA8 cells showed that maximum down-regulation was seen at 48 hours. B) Effect of TG2 gene silencing on in vitro HeyA8 invasive potential. Each experiment was performed in triplicate and repeated at least two times. C) Effect of TG2 silencing on HeyA8 cell viability was examined using various concentrations of docetaxel (0 to 50 nM). The number of viable cells remaining were determined after 72 hours. Error bars represent S.D. D) TG2 expression was assessed in HeyA8 tumors growing in the peritoneal cavity of nude mice following 2 doses of i.p. TG2 siRNA-DOPC using. Upper panel, Western blot. Lower panel, immunohistochemical peroxidase staining for TG2. All pictures were taken at original magnification x100.
In vivo silencing by TG2 specific siRNA-DOPC
Based on the evidence that the overexpression of TG2 in ovarian cancer cells is closely related to many aggressive tumor features, we investigated its potential as a therapeutic target. Based on our prior experience with in vivo gene silencing, we tested two doses of TG2 siRNA-DOPC in vivo. Nude mice bearing HeyA8 tumors were injected intraperitoneally twice 48 hours apart with a dose of TG2 siRNA-DOPC 3.5 μg/mouse (105 μg /kg) or 5.0 μg/mouse (150 μg /kg) and tumors were harvested 2, 4, and 6 days after injection. With the 5.0 μg dose, 70% decrease in TG2 levels was observed that persisted for at least 4 days (Figure 3D). TG2 expression began to return toward basal levels by 6 days after treatment. Similar results were noted with immunohistochemistry (Figure 3D). TG2 expression was not affected by non-specific control siRNA. Therefore, we selected the 5.0 μg (150 μg /kg) dose given twice weekly of TG2 siRNA-DOPC as the dosing schedule for subsequent therapy experiments.
In vivo therapy experiments using liposomal siRNA targeting TG2
The HeyA8, RMG2, and HeyA8-MDR ovarian cancer cells were selected for the therapy experiments because these lines have high level of TG2 expression. Seven days following tumor cell injection into the peritoneal cavity of nude mice, therapy was started according to five treatment groups: empty liposome, non-specific control siRNA-DOPC, control siRNA-DOPC in combination with docetaxel, TG2 siRNA-DOPC, and TG2 siRNA-DOPC plus docetaxel. The animals were sacrificed after 3 to 4 weeks of therapy and a necropsy was performed. In the HeyA8 model, treatment with control siRNA-DOPC plus docetaxel, and TG2 siRNA-DOPC alone was effective in reducing tumor weight (p=0.016 and 0.011, respectively; Figure 4A). However, the greatest efficacy was observed with TG2 siRNA-DOPC plus docetaxel (91% reduction in tumor weight, p=0.001). The combination therapy of TG2 siRNA-DOPC with docetaxel was more effective than control siRNA-DOPC plus docetaxel (p=0.006) or TG2 siRNA-DOPC alone (p=0.03). Given the high prevalence of resistance to chemotherapy in patients with ovarian cancer, we also examined the effects of TG2 silencing using the HeyA8-MDR model. As expected, docetaxel had no effect on tumor growth (Figure 4B). However, TG2 siRNA-DOPC plus docetaxel reduced tumor weight significantly (p=0.032). In the RMG2 model, treatment with TG2 siRNA-DOPC alone, and TG2 siRNA-DOPC plus docetaxel was effective in reducing tumor weight (p=0.029 and 0.001, respectively; Figure 4C). However, the greatest efficacy was observed with TG2 siRNA-DOPC plus docetaxel (86% reduction in tumor weight). The combination therapy of TG2 siRNA-DOPC with docetaxel was more effective than control siRNA-DOPC plus docetaxel (p=0.001) or TG2 siRNA-DOPC alone (p=0.008).
Figure 4.
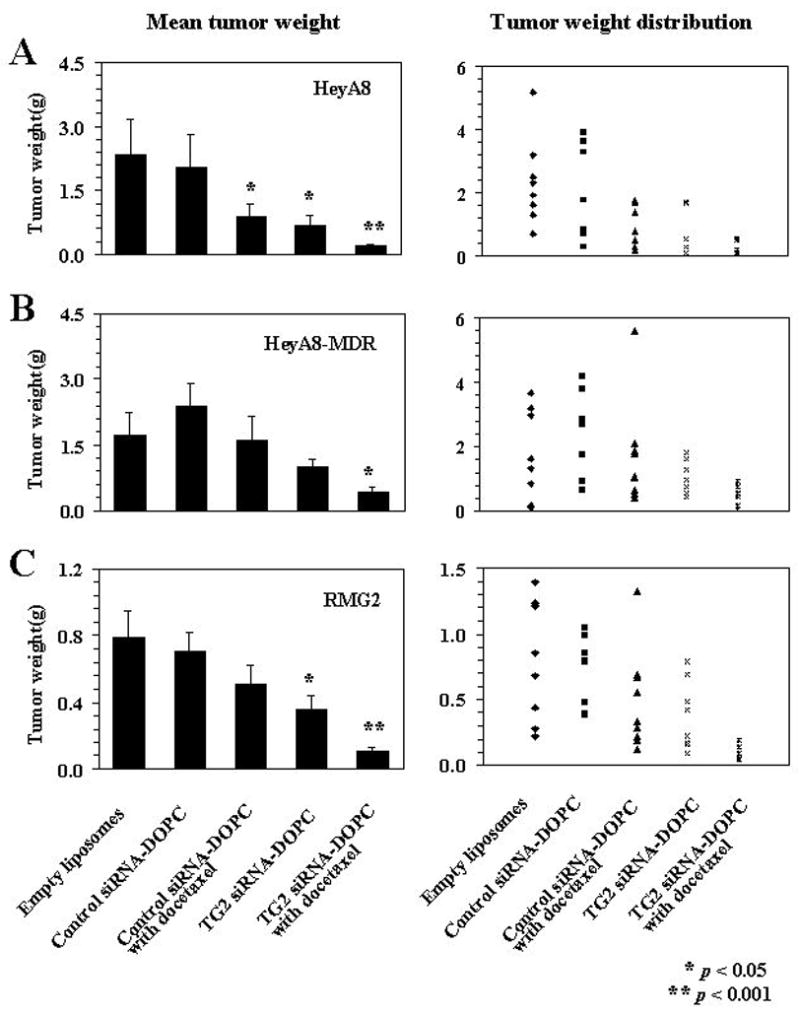
Therapeutic efficacy of TG2 siRNA with docetaxel. Nude mice were injected i.p. with HeyA8 (A) or HeyA8-MDR (B) or RMG2 (C) cells and randomly allocated to one of following groups (therapy beginning 1 week after tumor cell injection): empty liposomes, control siRNA-DOPC, control siRNA-DOPC with docetaxel, TG2 siRNA-DOPC, TG2 siRNA-DOPC with docetaxel. The animals were sacrificed when control mice became moribund (3–4 weeks after starting therapy) and necropsy was done. Error bars represent S.E.
Data from other measured variables of these therapy experiments are shown in Supplementary Table 3. The incidence of tumor formation was not significantly different among the five groups in either cell line. However, the number of nodules formed was reduced by treatment with control siRNA-DOPC plus docetaxel, TG2 siRNA-DOPC alone, and TG2 siRNA-DOPC plus docetaxel in HeyA8 cell injected mice (p values = 0.018, 0.013, 0.016, respectively) and RMG2 cell injected mice (p values = 0.015, 0.016, 0.002, respectively). We also examined TG2 expression at the conclusion of the therapy experiments by immunohistochemical staining analysis. TG2 expression levels were not affected by either control siRNA-DOPC, empty liposome, or control siRNA-DOPC plus docetaxel. However, in the TG2 siRNA-DOPC and combined TG2 siRNA-DOPC with docetaxel groups, there was sustained suppression of TG2 expression at the end of the therapy experiments (Figure 5A). There were no behavioral changes, such as change in eating habits or mobility in animals treated with liposomal siRNA preparations and mouse weights were not significantly different among the five groups of animals, suggesting that eating and drinking habits were not affected.
Figure 5.
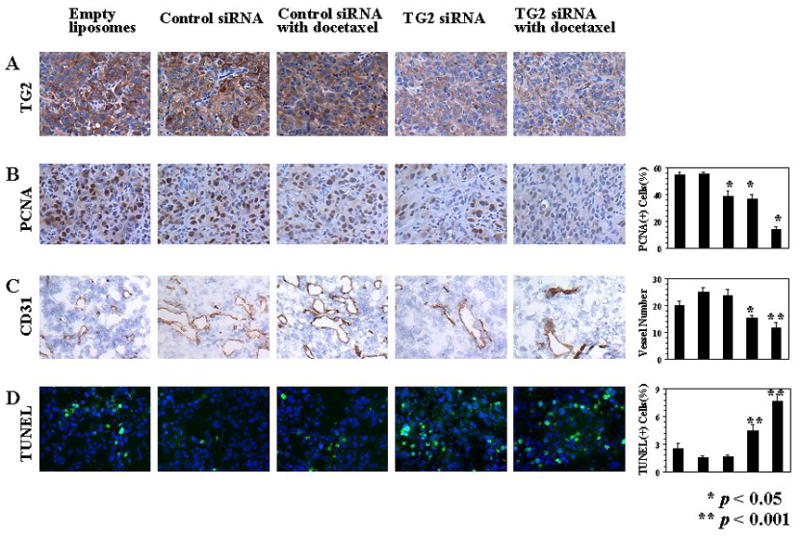
A) Effects of TG2-targeted therapy on TG2 expression (TG2; original magnification × 100); B) proliferation (PCNA; original magnification × 100); C) microvessel density (CD31; original magnification × 100), and D) apoptosis (green nuclei by TUNEL staining; blue nuclei by Hoechst; original magnification × 200). The graphs correspond to the labeled columns for immunohistochemistry. Error bars represent S.D.
Effect of TG2 targeting on cell proliferation, angiogenesis and apoptosis
To determine potential mechanisms underlying the efficacy of anti-TG2-based therapy, we examined its effects on several biological end-points, including proliferation (PCNA), angiogenesis (microvessel density; MVD), and apoptosis (TUNEL). First, we examined the effects of TG2-targeted therapy on tumor cell proliferation by using PCNA staining. Significant reduction of PCNA expression was observed in the groups treated with control siRNA-DOPC plus docetaxel, TG2 siRNA-DOPC alone, and TG2 siRNA-DOPC plus docetaxel (all p values < 0.01; Figure 5B). Next, we analyzed microvessel density (Figure 5C) in the harvested tumors using CD31 immunostaining. Compared with the empty liposome group, the mean MVD was significantly reduced in tumors treated with TG2 siRNA-DOPC alone and TG2 siRNA-DOPC plus docetaxel (p = 0.039 and 0.004, respectively). The most significant reduction in MVD occurred in the combination therapy group. To examine whether the TG2 siRNA-DOPC-mediated in vivo effects on endothelial cells could be direct, we treated murine endothelial cells isolated from the ovary of ImmortoMice [H-2k(b)-tsA58; ref. 28] and murine cancer cell lines with TG2 siRNA-DOPC. Murine TG2 levels were not altered by the TG2 siRNA-DOPC used for the in vivo experiments (data not shown). Finally, we evaluated tumor cell apoptosis by using TUNEL staining (Figure 5D). Minimal tumor cell apoptosis was apparent in either empty liposome, control siRNA-DOPC, or control siRNA-DOPC with docetaxel treatment groups, however, treatment with TG2 siRNA-DOPC alone and TG2 siRNA-DOPC plus docetaxel resulted in a significant increase in apoptosis (p = 0.027 and 0.001, respectively). Interestingly, the increase in apoptosis in the TG2 siRNA-DOPC plus docetaxel group was greater than the TG2 siRNA-DOPC alone group (p = 0.004).
Discussion
In this study, we found that overexpression of TG2 in ovarian carcinoma was significantly associated with worse overall patient survival. Moreover, TG2 promoted several biological functions of cancer cells such as cell attachment, invasion, and chemotherapy-resistance. Furthermore, our in vivo experiments indicate that treatment with TG2 siRNA-DOPC plus chemotherapy was highly effective in reducing ovarian cancer growth.
In epithelial ovarian cancer, the most frequent and earliest route of spread is by exfoliation of cells that implant along the surfaces of peritoneal cavity. Based on the adhesion assays, TG2 may play a role in the early steps of ovarian cancer metastasis. These findings are further supported by recent observations regarding diminished dissemination of tumors on the peritoneal surface and mesentery following TG2 knockdown in an i.p. ovarian xenograft model (29). This phenotype was associated with deficient integrin-fibronectin interaction, leading to weaker anchorage of cancer cells to the peritoneal matrix.
Drug resistance and metastasis are major impediments for the successful treatment of ovarian cancer. A common feature among drug resistant and metastatic tumor cells is that they exhibit profound resistance to apoptosis (30). Intracellular TG2 is able to cross-link the inhibitory subunit alpha of NF-κB (I-κBα) and, thus, constitutively activate the transcription factor nuclear factor--κB (NF-αB) which, in turn, promotes expression of anti-apoptotic proteins such as Bcl-xL and BFL1 (31, 32). Kim and colleagues reported that increased expression of TG2 and subsequent activation of NF-κB may contribute to drug resistance in breast cancer cells independently of EGF signaling (33). Consistent with these findings, in vivo TG2 silencing with siRNA enhanced tumor cell apoptosis in our therapy models.
Although TG2 silencing seems to have direct anti-tumor effects, there is growing evidence that the tumor microenvironment may also be affected. Recently, it was demonstrated that a plasma transglutaminase, thrombin-activated FXIII (FXIIIA-subunit), activates VEGFR-2 by cross-linking it with the αvβ3 integrin on the surface of endothelial cells, thereby stimulating angiogenesis (34). In the current study, we found that treatment with TG2 siRNA-DOPC plus docetaxel decreased the microvessel density in tumor tissue. Further work is needed to determine the impact of TG2 on tumor angiogenesis. Recently, one study showed that TG2 expression in pancreatic cancer cells induced constitutive activation of focal adhesion kinase (FAK) and its downstream PI3K/Akt survival pathway and it was independent of its enzymatic activity (19). Consistent with this, our in vitro experiments showed that overexpression of TG2 in SKOV3ip1 cells transfected with TG2-W induced increased phosphorylation of Akt. FAK is a non-receptor kinase that is a critical mediator of signaling between cells and their extracellular matrix (35). FAK activation at focal adhesion sites leads to cytoskeletal reorganization, cellular adhesion, and survival, and it is known to play a role in cell migration and invasion (22). In our previous experiments, FAK silencing was associated with lower levels of VEGF and matrix metalloproteinase-9 and increased apoptosis of tumor-associated endothelial cells, suggesting an anti-vascular effect. Therefore, TG2 may affect tumor angiogenesis indirectly by reducing FAK activation.
In another aspect, TG2 also plays certain roles in host protection and physiology. Several lines of evidence suggest that tissue transglutaminase plays an important role in stabilizing the ECM by cross-linking its component proteins and rendering it resistant to mechanical and proteolytic degradation (5, 9) and contributes to fibroblast wound healing processes (36). Furthermore, it was reported that TG2-induced alterations in the ECM of host could effectively inhibit the process of metastasis (37). Although TG2 silencing was very effective in treating ovarian cancer in our orthotopic models, we cannot fully determine the significance of tumor versus host TG2. Future studies using TG2-null background mice will be helpful in clarifying the role of host versus tumor TG2 in progression of ovarian cancer.
TG2 overexpression has been implicated in the pathogenesis of a number of medical diseases, such as celiac sprue (38), neurodegenerative disorders (39), diabetes (40), liver cirrhosis and fibrosis (41), and renal scarring (42) as well as various cancers. Although the current study was focused on ovarian cancer, TG2 silencing using siRNA incorporated in neutral liposomes may be useful for management of other benign and malignant diseases. Several kinds of TG2 inhibitors are being developed for disease control such as competitive amine inhibitors, reversible inhibitors, and irreversible inhibitors (43). Recently, some pre-clinical data with TG2 inhibitors have been reported. For example, treatment of glioblastoma cells in culture with the competitive TG2 inhibitor, monodansylcadarevine (MDC) or with the selective small molecule-irreversible TG2 inhibitor, KCA075, or its analog KCC009 showed an increased incidence of tumor cell apoptosis (44). In addition, KCC009 treatment in mice harboring orthotopic glioblastomas sensitized the tumors to N,N′-bis(2-chloroethyl)-N-nitrosourea chemotherapy, as measured by reduced bioluminescence, increased apoptosis and prolonged survival. Our systemic siRNA approach offers an attractive alternative option for therapeutic targeting of TG2.
In summary, targeted therapy with TG2 siRNA-DOPC in combination with chemotherapy significantly reduces tumor growth in both chemotherapy-sensitive and chemotherapy-resistant models. This anti-tumor effect was closely related to reduced proliferation, decreased angiogenesis, and increased tumor cell apoptosis, in addition to decreased attachment and invasion. Given the clinical relationship between TG2 and ovarian cancer prognosis, our findings raise the possibility that TG2 silencing in combination with docetaxel chemotherapy could be a novel therapeutic option against advanced ovarian carcinoma.
Supplementary Material
Acknowledgments
The authors thank Donna Reynolds and Dr. Robert Langley for assistance with immunohistochemistry and helpful discussion.
This research was funded in part by NIH grants (CA109298, CA110793, and CA092115), The Marcus Foundation, the U.T.M.D. Anderson Cancer Center SPORE in Ovarian Cancer (P50CA083639), a Program Project Development Grant from the Ovarian Cancer Research Fund, Inc., and the Zarrow Foundation.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA: a cancer journal for clinicians. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. The New England journal of medicine. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 3.Chandrashekar R, Tsuji N, Morales T, Ozols V, Mehta K. An ERp60-like protein from the filarial parasite Dirofilaria immitis has both transglutaminase and protein disulfide isomerase activity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(2):531–6. doi: 10.1073/pnas.95.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta K. Mammalian transglutaminases: a family portrait. Progress in experimental tumor research. 2005;38:1–18. doi: 10.1159/000084229. [DOI] [PubMed] [Google Scholar]
- 5.Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends in biochemical sciences. 2002;27(10):534–9. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 6.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nature reviews. 2003;4(2):140–56. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 7.Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. The Journal of biological chemistry. 2004;279(23):23863–8. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- 8.Baek KJ, Kang S, Damron D, Im M. Phospholipase Cdelta1 is a guanine nucleotide exchanging factor for transglutaminase II (Galpha h) and promotes alpha 1B-adrenoreceptor-mediated GTP binding and intracellular calcium release. The Journal of biological chemistry. 2001;276(8):5591–7. doi: 10.1074/jbc.M008252200. [DOI] [PubMed] [Google Scholar]
- 9.Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connective tissue research. 2000;41(1):1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 10.Milakovic T, Tucholski J, McCoy E, Johnson GV. Intracellular localization and activity state of tissue transglutaminase differentially impacts cell death. The Journal of biological chemistry. 2004;279(10):8715–22. doi: 10.1074/jbc.M308479200. [DOI] [PubMed] [Google Scholar]
- 11.Mishra S, Murphy LJ. The p53 oncoprotein is a substrate for tissue transglutaminase kinase activity. Biochemical and biophysical research communications. 2006;339(2):726–30. doi: 10.1016/j.bbrc.2005.11.071. [DOI] [PubMed] [Google Scholar]
- 12.Mishra S, Saleh A, Espino PS, Davie JR, Murphy LJ. Phosphorylation of histones by tissue transglutaminase. The Journal of biological chemistry. 2006;281(9):5532–8. doi: 10.1074/jbc.M506864200. [DOI] [PubMed] [Google Scholar]
- 13.Akimov SS, Belkin AM. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: a role in TGFbeta-dependent matrix deposition. Journal of cell science. 2001;114(Pt 16):2989–3000. doi: 10.1242/jcs.114.16.2989. [DOI] [PubMed] [Google Scholar]
- 14.Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006;11:1057–76. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
- 15.Singh US, Pan J, Kao YL, Joshi S, Young KL, Baker KM. Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. The Journal of biological chemistry. 2003;278(1):391–9. doi: 10.1074/jbc.M206361200. [DOI] [PubMed] [Google Scholar]
- 16.Fok JY, Ekmekcioglu S, Mehta K. Implications of tissue transglutaminase expression in malignant melanoma. Molecular cancer therapeutics. 2006;5(6):1493–503. doi: 10.1158/1535-7163.MCT-06-0083. [DOI] [PubMed] [Google Scholar]
- 17.Herman JF, Mangala LS, Mehta K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene. 2006;25(21):3049–58. doi: 10.1038/sj.onc.1209324. [DOI] [PubMed] [Google Scholar]
- 18.Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene. 2007;26(17):2459–70. doi: 10.1038/sj.onc.1210035. [DOI] [PubMed] [Google Scholar]
- 19.Verma A, Wang H, Manavathi B, et al. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer research. 2006;66(21):10525–33. doi: 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- 20.Landen CN, Jr, Chavez-Reyes A, Bucana C, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer research. 2005;65(15):6910–8. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 21.Halder J, Kamat AA, Landen CN, Jr, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res. 2006;12(16):4916–24. doi: 10.1158/1078-0432.CCR-06-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood AK, Coffin JE, Schneider GB, et al. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. The American journal of pathology. 2004;165(4):1087–95. doi: 10.1016/S0002-9440(10)63370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sood AK, Seftor EA, Fletcher MS, et al. Molecular determinants of ovarian cancer plasticity. The American journal of pathology. 2001;158(4):1279–88. doi: 10.1016/S0002-9440(10)64079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JS, Agarwal N, Mehta K. Multidrug-resistant MCF-7 breast cancer cells contain deficient intracellular calcium pools. Breast cancer research and treatment. 2002;71(3):237–47. doi: 10.1023/a:1014461832403. [DOI] [PubMed] [Google Scholar]
- 25.Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res. 2004;10(23):8068–76. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- 26.Landen CN, Merritt WM, Mangala LS, et al. Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer. Cancer biology & therapy. 2006;5(12):1708–13. doi: 10.4161/cbt.5.12.3468. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Vuori K, Reed JC, Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(13):6161–5. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langley RR, Ramirez KM, Tsan RZ, Van Arsdall M, Nilsson MB, Fidler IJ. Tissue-specific microvascular endothelial cell lines from H-2K(b)-tsA58 mice for studies of angiogenesis and metastasis. Cancer research. 2003;63(11):2971–6. [PubMed] [Google Scholar]
- 29.Satpathy M, Cao L, Pincheira R, et al. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer research. 2007;67(15):7194–202. doi: 10.1158/0008-5472.CAN-07-0307. [DOI] [PubMed] [Google Scholar]
- 30.Fok JY, Mehta K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis. 2007;12(8):1455–63. doi: 10.1007/s10495-007-0079-3. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Kim YS, Choi DH, et al. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. The Journal of biological chemistry. 2004;279(51):53725–35. doi: 10.1074/jbc.M407627200. [DOI] [PubMed] [Google Scholar]
- 32.Mann AP, Verma A, Sethi G, et al. Overexpression of Tissue Transglutaminase Leads to Constitutive Activation of Nuclear Factor-{kappa}B in Cancer Cells: Delineation of a Novel Pathway. Cancer research. 2006;66(17):8788–95. doi: 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- 33.Kim DS, Park SS, Nam BH, Kim IH, Kim SY. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-kappaB inactivation. Cancer research. 2006;66(22):10936–43. doi: 10.1158/0008-5472.CAN-06-1521. [DOI] [PubMed] [Google Scholar]
- 34.Dardik R, Inbal A. Complex formation between tissue transglutaminase II (tTG) and vascular endothelial growth factor receptor 2 (VEGFR-2): proposed mechanism for modulation of endothelial cell response to VEGF. Experimental cell research. 2006;312(16):2973–82. doi: 10.1016/j.yexcr.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochimica et biophysica acta. 2001;1540(1):1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 36.Stephens P, Grenard P, Aeschlimann P, et al. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. Journal of cell science. 2004;117(Pt 15):3389–403. doi: 10.1242/jcs.01188. [DOI] [PubMed] [Google Scholar]
- 37.Mangala LS, Arun B, Sahin AA, Mehta K. Tissue transglutaminase-induced alterations in extracellular matrix inhibit tumor invasion. Molecular cancer. 2005;4:33. doi: 10.1186/1476-4598-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molberg O, McAdam SN, Sollid LM. Role of tissue transglutaminase in celiac disease. Journal of pediatric gastroenterology and nutrition. 2000;30(3):232–40. doi: 10.1097/00005176-200003000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Hoffner G, Djian P. Transglutaminase and diseases of the central nervous system. Front Biosci. 2005;10:3078–92. doi: 10.2741/1764. [DOI] [PubMed] [Google Scholar]
- 40.Bernassola F, Federici M, Corazzari M, et al. Role of transglutaminase 2 in glucose tolerance: knockout mice studies and a putative mutation in a MODY patient. Faseb J. 2002;16(11):1371–8. doi: 10.1096/fj.01-0689com. [DOI] [PubMed] [Google Scholar]
- 41.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126(7):1795–808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Johnson TS, El-Koraie AF, Skill NJ, et al. Tissue transglutaminase and the progression of human renal scarring. J Am Soc Nephrol. 2003;14(8):2052–62. doi: 10.1097/01.asn.0000079614.63463.dd. [DOI] [PubMed] [Google Scholar]
- 43.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacology & therapeutics. 2007;115(2):232–45. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan L, Siegel M, Choi K, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26(18):2563–73. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.