

Abstract
Dynamic light scattering and Fourier transform infrared spectroscopy were used to study the formation of prefibrillar aggregates and fibrils of bovine pancreatic insulin at 60°C and at pH 1. The kinetics of disintegration of the prefibrillar aggregates were also studied using these techniques after a quench to 25°C. These experiments reveal that formation of prefibrillar aggregates is reversible under the solution conditions studied and show that it is possible to significantly reduce the nucleation (lag) times associated with the onset of fibril growth in bovine pancreatic insulin solutions by increasing the concentration of prefibrillar aggregates in solution. These results provide convincing evidence that less structured prefibrillar aggregates can act as fibril-forming intermediates.
INTRODUCTION
Amyloid fibril formation is increasingly being considered to be a property of all globular proteins (1). A recent article by Chiti and Dobson has cited 40 different human diseases that have been linked with the formation of these amyloid protein deposits (2). However, fibrils are not the only type of protein aggregate that have been associated with these conditions. A number of recent studies have also reported the existence of smaller aggregates that precede fibril formation (3–9). The importance of understanding the processes that are involved in the formation of the aggregates that precede fibril formation (prefibrillar aggregates) has been highlighted because of evidence that they (and not mature fibrils) may be the main cytotoxic species (6,8–11). It has been suggested that it is the misfolded nature of the proteins that leads to the cytotoxicity of these aggregates (8). Misfolded globular proteins often have exposed hydrophobic residues that are usually hidden in the core of the molecules. The exposure of these residues could be responsible for abnormal interactions between the protein molecules and the constituents of a cell. As a result, the biochemical reactions that take place in and around the cell may be hindered by the presence of these aggregates (2). However, recent work by Yoshiike et al. (12) has challenged these ideas. These authors propose that the lack of correlation that has been reported between the quantity of fibrils and their measured toxicity could be caused by variations in the physicochemical properties of different fibril morphologies.
Smith et al. studied insulin fibril formation and showed that small aggregates with an average radius of ∼13 nm formed before the fibrils (13). The insulin molecules that made up these prefibrillar aggregates were found to adopt a conformation containing a small amount of intermolecular β-sheet structure. Ahmad et al. (7) have also provided experimental evidence for the existence of prefibrillar aggregates at several lower protein concentrations than those studied by Smith and co-workers. These studies indicate that prefibrillar aggregates are found under a variety of solution conditions that are capable of initiating fibril formation.
A key question that arises from these studies is related to whether prefibrillar aggregates are a necessary precursor for the nucleation of fibrils, or whether they simply represent a competing aggregation pathway. Studies have been published that indicate that both of these scenarios can occur (4,5,14–16). The task of elucidating the relationship between prefibrillar aggregates and protein fibril nucleation therefore continues to be of considerable importance.
In this article, we describe an experimental study of the formation of prefibrillar aggregates and fibrils in solutions of bovine pancreatic insulin (BPI) at elevated temperature and at pH 1. A combination of Fourier transform infrared (FTIR) spectroscopy and dynamic light scattering (DLS) measurements were used to study the formation of these aggregates at 60°C. These techniques were also used to study the disintegration of prefibrillar aggregates after a rapid temperature quench from 60°C to 25°C. We show that it is possible to vary the concentration of prefibrillar aggregates in solution by varying the time after the quench. We also show that the nucleation (lag) time associated with the formation of fibrils depends upon the concentration of prefibrillar aggregates in solution. These experiments provide convincing evidence that the presence of nonfibrillar aggregates in solution can reduce the fibril nucleation (lag) times and promote the onset of insulin fibril formation.
METHODS
Protein sample preparation
Stock solutions containing 0.025 M NaCl and 0.1 M HCl (pH 1) were prepared in preboiled deuterium oxide (D2O; Goss Scientific Instruments, Chelmsford, Essex, UK). These solutions were sealed in 15 mL sample vials and degassed by placing them in a temperature-controlled water bath at 85°C for ∼1 h. The solutions were then allowed to cool to room temperature. Bovine pancreatic insulin (BPI; Mw = 5733, cat. No. I5500, Sigma Aldrich, Gillingham, Dorset, UK) was dissolved in the stock solutions at a concentration of 20 mg mL−1 (∼3.5 mM). The resulting protein solutions were left at room temperature in sealed vials for a further 24 h to ensure that all the protein had dissolved.
After the protein had dissolved, the sealed sample vials were heated to 60°C on a THMS600 hot-stage (Linkam Scientific Instruments, Tadworth, Surrey, UK) using a homebuilt heater assembly. The vials were held at this temperature for 1 h. This procedure has been shown to result in the formation of prefibrillar aggregates in the protein solutions, but no insulin fibrils are formed on these timescales (13). The samples were then quenched rapidly to 25°C by placing them in a temperature-controlled water bath.
The protein solutions were then separated into two sets (A and B, respectively) and each sample set was subjected to a different thermal history. Sample set A was reheated to 60°C immediately (within 5 min) after the quench and was maintained at this temperature to allow the aggregation/fibril formation of the protein to reach completion. Sample set B was maintained at a temperature of 25°C for 120 h after the quench. This protein solution was then heated to 60°C and allowed to undergo further aggregation and fibril formation. In each case, the samples were held at 60°C for at least 150 min after being reheated. Previous work has shown that this is the length of time required for the aggregation of BPI to reach completion at this temperature and under this set of solution conditions (13).
In all the experiments described below, duplicate measurements were carried out. Additional experiments could not be performed, as all the experiments were carried out using a single batch of insulin. This was done because significant batch-to-batch variations in the properties of insulin have been reported (13,17). The use of a single batch of BPI therefore ensures that any observed changes in the kinetics of aggregation of the samples are entirely due to differences in the way the samples are treated and not due to the batch dependent properties of the protein.
Dynamic light scattering
Small aliquots of BPI solution were taken from sample B at different times after the temperature quench from 60°C to 25°C. These aliquots were rapidly transferred to a quartz cuvette and DLS measurements were performed using a Viscotek (Crowthorne, Berkshire, UK) DLS apparatus (laser wavelength 633 nm). Before collection of the experimental data, the DLS equipment was calibrated against a series of polystyrene colloid standards of different sizes (46 nm, 98 nm, and 306 nm). The temperature was maintained at 25°C throughout the experiments and the size distributions of the aggregates and the total scattered light intensity were recorded as a function of time. Data were initially collected at 5-min intervals for a period of 2 h. Following this, data were collected at 1 h intervals for a further 21 h.
Fourier transform infrared spectroscopy
Aliquots of solutions taken from sample B were also injected into an FTIR liquid transmission cell (75-μm pathlength; Specac, Orpington, Kent, UK) after the temperature quench. The temperature of the cell was maintained at 25°C and infrared spectra were collected every 5 min (averaging 250 scans per spectrum, 4 cm−1 resolution) for a period of 30 h using an FTS40 Pro spectrometer (Varian, Oxford, UK) equipped with Resolutions Pro 4.0 software (Varian, Oxford, UK). All the spectra collected were ratioed to a background spectrum (averaging 250 scans per spectra) of the sample solution that did not contain BPI.
FTIR spectra were also collected from samples A and B and used to follow the aggregation process by monitoring changes in the β-sheet content of the BPI solutions when the samples were reheated to 60°C. During these experiments, infrared spectra were collected at 15-s intervals for a period of 150 min. All spectra were collected using a resolution of 4 cm−1.
RESULTS AND DISCUSSION
Fig. 1 shows size distributions obtained from DLS measurements of the BPI solutions that were collected at different times after a temperature quench from 60°C to 25°C. All the data shown in this figure is normalized so that the intensity of the peak corresponding to the largest particle size observed is equal to one. This allows a qualitative comparison of the relative concentrations of each aggregate type to be made at each time. Fig. 1 shows that shortly after the quench, the BPI solutions contain a significant number of aggregates with an average radius of ∼13 nm as well as a small number of structures that have a radius of ∼2–4 nm. This figure also shows that the distribution of sizes of the larger (∼13 nm) aggregates varies between 7 nm and 30 nm. The smaller (2–4 nm) structures are similar in size to those observed when insulin is dissolved at 25°C under the solution conditions used in this study (pH 1, 0.025 M NaCl). They are comparable to structures reported in other studies of insulin and are likely to arise as a result of the formation of insulin dimers and larger oligomeric structures that are known to occur under these solution conditions (7,18).
FIGURE 1.

Particle size distributions obtained from DLS studies of prefibrillar aggregates in solutions of BPI (20 mg mL−1, pH 1) after a temperature quench from 60°C to 25°C. Data were collected at times of 150 min (dotted line), 500 min (dashed line), and 2940 min (solid line) after the quench. All size distributions are normalized in such a way that the size of the peak centered at ∼13 nm is equal to unity.
The larger (13 nm) prefibrillar aggregates were observed to disintegrate into the ∼2–4 nm aggregates after the quench to 25°C. This can be observed in Fig. 1 by the increase in the relative size of the peak corresponding to the 2–4 nm aggregates. It is worth noting that none of the DLS size distributions collected during these experiments revealed the presence of any aggregates of intermediate or larger sizes. The upper limit of the length scales that can be measured using DLS is ∼1 μm. Aggregates that are ≥1 μm in size would be clearly visible in solution. No structures of this size were observed in the solutions containing the prefibrillar aggregates. However, when the solutions were incubated at 60°C for extended periods and fibrils were allowed to form, the solutions were observed to go cloudy. These observations are consistent with reports obtained in a previous study in which insulin fibrils were formed under similar solution conditions (13).
A more detailed inspection of the size distributions shown in Fig. 1 reveals that there is some evidence for a narrowing in the distributions of the larger (∼13 nm) aggregate sizes as they disintegrate. It is not clear whether the narrowing is related to some property of the size-dependent disintegration kinetics of the aggregates or it is simply an artifact of the assumptions that are made about the geometry of the aggregates when calculating the size distributions (see below). Determining the reasons for the narrowing of the distribution of sizes with increasing disintegration times would be difficult without a more detailed (real or reciprocal space) analysis of the size distributions of the aggregates using a number of different experimental techniques. Such an analysis is beyond the scope of this study.
The kinetics of prefibrillar aggregate disintegration can be followed more clearly by monitoring the total intensity of light scattered from all the objects in solution as a function of time (5). Fig. 2 shows the time-dependent decay in the total scattering intensity measured in the DLS experiments after a quench from 60°C to 25°C. This figure shows that as time proceeds and the larger aggregates disintegrate into smaller structures, the total scattering intensity decreases. These measurements also indicate that the decay in scattering intensity and hence the dissociation of the prefibrillar aggregates takes place on timescales in excess of 40 h.
FIGURE 2.

Light scattering and FTIR studies of the disintegration of prefibrillar aggregates in solutions of BPI (20 mg mL−1, pH 1) after a temperature quench from 60°C to 25°C. Data are shown for the decay in the total light scattering intensity (▪, DLS) and the area of the ratioed β-sheet peak (○, FTIR, see text) as a function of time. The inset shows an AFM image of clusters of prefibrillar aggregates taken over a 5 μm × 5 μm area. The scale bar on this image corresponds to a distance of 1 μm.
The observed decay in the scattering intensity can be interpreted using basic Rayleigh scattering theory. If we assume that to a first approximation, all the scattering objects in solution can be treated as spheres, the scattering intensity would be expected to scale as ∼cR6 (where R and c are the radius and concentrations of the aggregates, respectively) (19). During the disintegration of the larger aggregates, the concentration of smaller aggregates would be expected to increase by a factor of 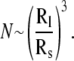 As a result, the scattering intensity from a population comprised entirely of smaller aggregates (Is) would be related to the intensity from a population of the larger aggregates (Il) by the equation
As a result, the scattering intensity from a population comprised entirely of smaller aggregates (Is) would be related to the intensity from a population of the larger aggregates (Il) by the equation
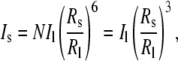 |
(1) |
where Rs and Rl are the radii of the small and large aggregates, respectively. Equation 1 confirms that the total scattering intensity is expected to decrease as the larger aggregates disintegrate into the smaller aggregate structures.
The assumption that the aggregates are spherical is likely to be a poor one. Evidence for this was obtained by using an Asylum Research MFP 3D atomic force microscope (AFM, operating in tapping mode) to collect images of dried solutions that contain clusters of prefibrillar aggregates. As shown in the inset of Fig. 2, the larger aggregates are not spherical objects. There is some indication that the prefibrillar aggregates are relatively compact but that they have a slightly elongated structure. This will modify the interpretation of the light scattering intensity data shown in Fig. 2, because a change in the geometry of the scattering objects will introduce a different size dependence into the scattering intensity. However, the qualitative behavior of the size dependence of the scattering intensity would be expected to be the same (i.e., a decrease in scattering intensity with decreasing object size). It is also worth noting that the DLS software used to determine the size of the aggregates in solution makes the assumption that the scattering objects are spherical. As discussed above, this assumption may not be valid for the larger aggregates and great care must be taken when interpreting the DLS size distributions shown in Fig. 1. As a result, the value of ∼13 nm represents an average measure of the hydrodynamic radius of the larger aggregates.
A more detailed examination of the AFM image shown in the inset of Fig. 2 shows that the lateral sizes of the structures observed are significantly larger than the ∼13 nm radius aggregates detected in the DLS measurements. The reason for this is that the image shown in the inset of Fig. 2 was obtained from a sample that was prepared by drying the BPI solutions on a clean polystyrene-coated, single crystal silicon wafer. During drying of the solutions, the aggregates cluster together under the influence of surface tension at the receding edge and surface of the drying droplet. Care must therefore be taken when interpreting AFM images such as the one shown in the inset of Fig. 2. AFM images of dried samples can only be used to obtain a qualitative comparison of the aggregate morphologies that exist in a given sample (13). However, it is worth noting that AFM line scans taken across the aggregate clusters shown in the image in Fig. 2 reveal that the height of these objects is 8 ± 1 nm. This value is comparable to the ∼13-nm average hydrodynamic radius of the aggregates that was obtained from the DLS measurements.
The disintegration of the prefibrillar aggregates was also studied using FTIR spectroscopy. This technique enables changes in the secondary structure of the protein that are associated with the state of aggregation to be monitored as a function of time (13). Fig. 3 shows FTIR spectra of the BPI solutions taken at times of 5, 60, 150, 500, and 2100 min after the quench from 60°C to 25°C. This figure shows that as the prefibrillar aggregates disintegrate, there is also a considerable change in the shape of the Amide I region of the IR spectrum of insulin. As discussed in a previous publication (13), this region can be accurately fitted to three Lorentzian peaks which correspond to vibrational frequencies associated with random coil (∼1645 cm−1), α-helix (∼1658 cm−1), and β-sheet structures (∼1610–1633 cm−1), respectively. Fitting Lorentzian line shapes to these peaks enables the relative content of these structures within the protein molecules to be determined (13). Fig. 3 shows that there is a gradual decrease in the size of the β-sheet shoulder (∼1620 cm−1) of the amide I region of the protein spectra with time, after the quench to 25°C. Qualitatively, this suggests that the β-sheet content of the protein molecules that comprise the ∼13-nm aggregates decreases as the aggregates disintegrate.
FIGURE 3.

FTIR spectra collected during the disintegration of prefibrillar aggregates in solutions of BPI (20 mg mL−1, pH 1) after a temperature quench from 60°C to 25°C. Data are shown for spectra collected at times of 5 (○), 60 (□), 150 (▵), 500 (•), and 2100 min (▪) after the quench. The solid line is a spectrum taken from freshly dissolved insulin at 25°C.
A more detailed analysis of the relative areas of the peaks corresponding to the different secondary structures (α-helix, β-sheet, and random coil) was performed on the protein molecules in the BPI solutions. Table 1 gives a summary of the relative proportions of the different secondary structures obtained from the IR spectra of the disintegrating aggregates of BPI shown in Fig. 3. The relative proportions of the different secondary structures obtained from a freshly dissolved BPI solution are also shown in this table for comparison. As shown in Table 1, the ratio of (β-sheet:α-helix/random coil) in the protein varied from 32%:33%:35% (immediately after the quench to 25°C) to 10%:53%:37% after ∼35 h. These values confirm that the disintegration of the larger prefibrillar aggregates is accompanied by significant changes in the secondary structure of the protein. These changes are consistent with other reports of differences in the secondary structure of prefibrillar aggregates and freshly dissolved insulin that were collected under similar solution conditions (13,20,21).
TABLE 1.
| % Content
|
|||
|---|---|---|---|
| Time (min) | β-sheet | Random coil | α-Helix |
| 5 | 32 ± 1 | 35 ± 1 | 33 ± 1 |
| 60 | 28 ± 1 | 38 ± 1 | 34 ± 1 |
| 150 | 23 ± 1 | 40 ± 1 | 37 ± 1 |
| 500 | 19 ± 1 | 39 ± 1 | 42 ± 1 |
| 2100 | 10 ± 1 | 37 ± 1 | 53 ± 1 |
| Freshly dissolved | 7 ± 1 | 39 ± 1 | 54 ± 1 |
Time dependence of the secondary structure content of BPI solutions after the quench from 60°C to 25°C. Data were obtained by calculating the areas of three Lorentzian line shapes (corresponding to β-sheet, α-helix, and random coil, respectively) that were fitted to each of the spectra shown in Fig. 3 (see text).
The peak fitting procedure employed here (and described in detail elsewhere (13)) was used to determine the variation in the β-sheet content of the protein as a function of time (see Fig. 2). The area of the β-sheet peak plotted in Fig. 2 has been divided by the total area of the amide I region. The total area of the amide I region contains information about all of the secondary structures that exist within the protein molecules in solution and hence provides a quantitative measure of the amount of protein in the beam path. Dividing the area of the β-sheet peak by the total area of the amide I peak in this way therefore provides a measure of the amount of β-sheet per unit volume in the samples being studied (13).
Fig. 2 shows that the kinetics associated with the decrease in β-sheet content of BPI occur on similar timescales to the disintegration of the larger aggregates (as determined from the decrease in light scattering intensity). As the prefibrillar aggregates disintegrate, the shape of the amide I peak (and hence the relative proportions of the different secondary structures) appears to revert to a state that is similar to that observed in freshly dissolved insulin (see Figs. 2 and 3 and also Table 1). The correspondence between the timescales associated with the β-sheet content of BPI molecules and the dissociation of prefibrillar aggregates (measured by DLS) suggests that intermolecular β-sheet plays an important role in determining the structure of the prefibrillar aggregates reported here (as has been established for the case of protein fibrils (18)). However, as shown previously, the β-sheet content per unit volume of the prefibrillar aggregates is smaller than that observed for insulin fibrils (13).
Smith et al. (13) have also shown that prefibrillar aggregates of insulin are able to bind Thioflavin-T (ThT) dye molecules in such a way as to produce a significant amount of fluorescence. The presence of a measurable ThT fluorescence intensity (above background levels) suggests that these aggregates contain repeat β-sheet units that are aligned in such a way that the ThT dye molecules can lie across several β-strands associated with different protein molecules (22). This indicates that the structures formed by the insulin molecules within these aggregates display some of the character of molecules that have been incorporated into mature fibrils (7).
The ability to change the concentration of prefibrillar aggregates by varying the time after a temperature quench provides a useful way of assessing their importance for fibril formation. As described above, samples A and B were held at 25°C for different periods of time (5 min and 120 h, respectively) after a quench from 60°C. These samples were then reheated and incubated at 60°C to allow fibrils to form in solution. The differences in prefibrillar aggregate concentrations between these two solutions were then used to determine how prefibrillar aggregates influence the nucleation (lag) times associated with fibril formation. During this time, the β-sheet content of the samples was monitored using FTIR spectroscopy (see Methods). As shown by Smith et al., monitoring the β-sheet content of the protein molecules in solution provides a valid method of following the aggregation process in solutions of BPI (13).
Fig. 4 shows the variation of the β-sheet content of BPI molecules in samples A and B as a function of time. This figure also shows the variation in the β-sheet content of BPI taken from a sample of freshly dissolved protein that was incubated at 60°C for the entire period shown on the plot (i.e., with no temperature quench to 25°C). All of the plots shown in Fig. 4 display the characteristic two-phase aggregation kinetics that have been reported previously for BPI under these solution conditions (13). The region in the β-sheet formation kinetics up to the end of the first plateau corresponds to changes associated with the formation and saturation of a population of ∼13-nm prefibrillar aggregates. The region of growth after this plateau has been attributed to changes in the β-sheet content of the protein associated with the formation of fibrils (13). The horizontal error bars on the plots shown in Fig. 4 were determined from repeat measurements of the FTIR β-sheet formation kinetics and are a measure of the level of experimental uncertainty in the fibril nucleation (lag) times.
FIGURE 4.

Fibril nucleation studies in BPI solutions (20 mg mL−1, pH 1) with different thermal histories. Data are shown for the ratioed β-sheet growth kinetics obtained from FTIR spectroscopy measurements of BPI solutions (see text). The data were collected from freshly dissolved BPI solutions that were incubated at 60°C (•), as well as solutions that were heated to 60°C for 60 min before being quenched to 25°C and then reheated to 60°C within 5 min (▵) and held at 25°C for 120 h before being reheated to 60°C (○). The solid lines shown in this figure mark the position of the plateau corresponding to the end of prefibrillar aggregate formation and the slopes of the regions corresponding to the period of fibril growth (13), respectively. The nucleation (lag) times for fibril growth can be determined from the times at which the fibril growth slopes intersect the plateau line. The horizontal error bars represent the uncertainty in the fibril nucleation times obtained in each set of experiments and are shown on a single data point in each set for reasons of clarity.
The times associated with the onset of fibril nucleation for the two samples studied (A and B) are clearly different within the limits of experimental uncertainty (see Fig. 4). The nucleation time for the sample containing a significant population of ∼13 nm prefibrillar aggregates (sample A) is significantly shorter (40 ± 19 min) than the nucleation time observed in the samples where these aggregates have been partially dissolved by holding the protein solutions at 25°C for 120 h (sample B). These simple experiments indicate that the difference in time that the samples were kept at 25°C (and hence the size of the population of prefibrillar aggregates), before being reheated, affects fibril nucleation times.
The data shown for sample A in Fig. 4 clearly demonstrates that the fibril nucleation time is shortened by 52 ± 19 min relative to that observed for a solution of freshly dissolved protein incubated at the same temperature. As this time difference is the same within error as the 60 min that sample set A was incubated before the quench, these results indicate that rapidly quenching the protein solutions and then immediately reheating them preserves most of the population of the prefibrillar aggregates. As a result, the aggregation continues as if the quench had not occurred. However, when prefibrillar aggregates are allowed to dissociate (as in the case of sample B), the aggregation history of the system is effectively erased and the protein returns to a conformation which is predominantly α-helix and random coil (see Fig. 3). Whether the BPI molecules return to the same conformation as freshly dissolved insulin is not certain. However, the conformation contains similar proportions of the different secondary structures to those observed for freshly dissolved BPI (see Table 1), making this a reasonable assumption.
As mentioned above, the difference in fibril nucleation times observed between sample sets A and B was found to be 40 ± 19 min. It might be expected that if the protein in sample B had reverted to its native conformation, the difference in nucleation times would correspond closely to the 60 min of heating that each sample experienced before the FTIR transmission measurements (as observed for freshly dissolved insulin). However, while the difference in the nucleation times between sample sets A and B is comparable to 60 min (within the limits of experimental uncertainty), it should be noted that it is unlikely that the prefibrillar aggregates in sample set B would have completely dissociated, even after 120 h.
An alternative interpretation of the results shown in Fig. 4 could be given in terms of the differences in concentrations of the smaller protein structures (i.e., insulin monomers and dimers) that exist between the two samples. However, this is unlikely to be the dominant factor in affecting the fibril nucleation times. We would expect that decreasing the concentrations of these structures would reduce the probability of them interacting in solution. If these structures were directly responsible for nucleating fibrils, this reduction in the collision probability would result in a longer fibril nucleation time being observed in the solutions with the lower concentrations of these smaller protein structures (i.e., in sample A). This is not observed in the experiments reported here.
A consideration of Fig. 4 shows that once prefibrillar aggregates have formed, a further ∼80 min is required for fibril nucleation to take place. This can be deduced from the length of the plateau region observed in the first stage of the β-sheet formation kinetics shown for freshly dissolved insulin in Fig. 4. If prefibrillar aggregates are responsible for nucleus formation, as these experiments suggest, then why don't fibrils nucleate immediately after these aggregates have formed, and what processes are occurring during the period corresponding to the plateau region in the kinetics?
Closer inspection of the plateau in the FTIR β-sheet kinetics in Fig. 4 reveals that the plateau is not completely flat, but exhibits a slight increase throughout the time preceding fibril nucleation. These changes indicate that the physical environment of the β-strands of the BPI molecules within the prefibrillar aggregates is changing during this time. This suggests that some local rearrangement of the BPI molecules may be taking place within the aggregates and that some BPI molecules reorganize into conformations that are more conducive to fibril formation.
A number of previous experimental studies have also shown that less well-defined aggregates form before fibrils in other protein systems. These studies have shown that structural rearrangements of the protein molecules can take place within the prefibrillar aggregates and that these structural changes typically occur on timescales >∼10 h (2,4,5,10,14). For example, Petty et al. performed studies of the Syrian hamster prion peptide (H1) and used isotope-modified IR spectroscopy to show that before fibril growth there is a necessary reorganization and alignment of β-strands within prefibrillar aggregates (10). They showed that the rearrangement and alignment of the β-strands in these aggregates displayed an Arrhenius temperature dependence and that the structural changes lead to fibril nucleus formation. These combined results suggest that the time taken for realignment of β-strands within the prefibrillar aggregates is likely to be the rate-limiting step for fibril nucleation in the current studies and may explain the associated lag time after the saturation of the population of prefibrillar aggregates.
In summary, our results suggest that the prefibrillar aggregates formed in this study cause the insulin molecules to cluster together in relatively compact and slightly elongated structures. Our experiments indicate that prefibrillar aggregation involves the formation of some intermolecular β-sheet between the insulin molecules, but that the β-sheet content per unit volume of these aggregates is initially smaller than that of mature fibrils. However, the formation of prefibrillar aggregates does not appear to quench in the structure of the protein molecules, as there is some evidence of changes in the β-sheet content of the protein during the plateau region of the FTIR β-sheet kinetics and before the onset of fibril formation. This suggests that there is some annealing of the intermolecular β-sheet structure within these aggregates (see Fig. 4) and that the protein molecules have some freedom to sample different conformational states.
The experimental observation that prefibrillar aggregates reduce the fibril nucleation (lag) times provides strong evidence that these aggregates can play an important role in initiating fibril formation. One possible explanation for the reduction in fibril nucleation times could be given in terms of the high local concentrations of BPI molecules that exist within the prefibrillar aggregates. The radius of gyration of isolated insulin molecules under the solution conditions used in this study is ∼1.6 nm (23). If we assume that the molecules are closely packed within the ∼13-nm prefibrillar aggregates, then each aggregate would contain ∼500 molecules (depending on its size and shape). The localization of the protein molecules in these aggregates would be expected to increase the frequency associated with two given BPI molecules interacting and adopting the conformations required for fibril formation (when compared to isolated molecules and dimers in solution). This provides a possible explanation for the reason why the formation of prefibrillar aggregates reduces the nucleation times associated with fibril formation. However, we stress that determining the mechanism by which these aggregates promote the nucleation of fibrils is not possible without a detailed analysis of the molecular rearrangements that take place within the aggregate. Such an analysis is currently beyond the scope of this work.
CONCLUSIONS
Dynamic light scattering and FTIR spectroscopy were used to study the formation and disintegration of prefibrillar aggregates in solutions of bovine pancreatic insulin (BPI) at 60°C and pH 1. Prefibrillar aggregates of BPI were found to have an average radius of ∼13 nm and their formation was shown to be reversible. The protein molecules in the prefibrillar aggregates contained a greater amount of β-sheet content per unit volume than freshly dissolved insulin, but a lower β-sheet content than fibrils. The concentration of prefibrillar aggregates was shown to depend upon the thermal history of the samples. Solutions with high concentrations of prefibrillar aggregates were shown to display significantly shorter fibril nucleation (lag) times than freshly dissolved BPI and solutions containing fewer aggregates. These experiments suggest that prefibrillar aggregates act as nucleating agents for fibrils and that the localization of many (∼500) protein molecules in these aggregates enables the BPI molecules to undergo the structural rearrangements that are required for fibrillogenesis.
Acknowledgments
We thank Dr. Sno Stolnik (School of Pharmacy, University of Nottingham) for providing access to the dynamic light scattering equipment. We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, UK) and the University of Nottingham's Interdisciplinary Doctoral Training Centre in Nanoscience for funding this work.
Editor: Heinrich Roder.
References
- 1.Chiti, F., P. Webster, N. Taddei, A. Clark, M. Stefani, G. Ramponi, and C. Dobson. 1999. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA. 96:3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiti, F., and C. Dobson. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75:333–366. [DOI] [PubMed] [Google Scholar]
- 3.Walsh, D. M., D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. M. Condron, A. Lomakin, G. B. Benedek, D. J. Selkoe, and D. B. Teplow. 1999. Amyloid β-protein fibrillogenesis. J. Biol. Chem. 274:25945–25952. [DOI] [PubMed] [Google Scholar]
- 4.Serio, T. R., A. G. Cashikar, A. S. Kowal, G. J. Sawacki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf, and S. L. Lindquist. 2000. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 289:1317–1321. [DOI] [PubMed] [Google Scholar]
- 5.Souillac, P. O., V. N. Uversky, and A. L. Fink. 2003. Structural transformations of oligomeric intermediates in the fibrillation of the immunoglobin light chain LEN. Biochemistry. 42:8094–8104. [DOI] [PubMed] [Google Scholar]
- 6.Kayed, R., Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall, and C. G. Glabe. 2004. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad, A., V. N. Uversky, D. Hong, and A. L. Fink. 2005. Early events in the fibrillation of monomeric insulin. J. Biol. Chem. 280:42669–42675. [DOI] [PubMed] [Google Scholar]
- 8.Williams, A. D., M. Sega, M. Chen, I. Kheterpal, M. Geva, V. Berthelier, D. T. Kaleta, K. D. Cook, and R. Wetzel. 2005. Structural properties of Aβ protofibrils stabilized by a small molecule. Proc. Natl. Acad. Sci. USA. 102:7115–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayed, R., E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman, and C. G. Glabe. 2006. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 300:486–489. [DOI] [PubMed] [Google Scholar]
- 10.Petty, S. A., and S. M. Decatur. 2005. Intersheet rearrangement of polypeptides during nucleation of β-sheet aggregates. Proc. Natl. Acad. Sci. USA. 102:14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Austen, B. M., K. E. Paleologou, S. A. E. Ali, M. M. Qureshi, D. Allsop, and O. M. A. El-Agnaf. 2008. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer's β-amyloid peptide. Biochemistry. 47:1984–1992. [DOI] [PubMed] [Google Scholar]
- 12.Yoshiike, Y., T. Akagi, and A. Takashima. 2007. Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry. 46:9805–9812. [DOI] [PubMed] [Google Scholar]
- 13.Smith, M. I., J. S. Sharp, and C. J. Roberts. 2007. Nucleation and growth of insulin fibrils in bulk solution and at hydrophobic polystyrene surfaces. Biophys. J. 93:2143–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khurana, R., J. R. Gillespie, A. Talapatra, L. J. Minert, C. Ionescu-Zanetti, I. Millet, and A. L. Fink. 2001. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry. 40:3525–3535. [DOI] [PubMed] [Google Scholar]
- 15.Gosal, W. S., I. J. Morten, E. W. Hewitt, D. A. Smith, N. H. Thomson, and S. E. Radford. 2005. Competing pathways determine fibril morphology in the self-assembly of β2-microglobulin into amyloid. J. Mol. Biol. 351:850–864. [DOI] [PubMed] [Google Scholar]
- 16.Powers, E. T., and D. L. Powers. 2008. Mechanisms of protein fibril formation: nucleated polymerization with competing off-pathway aggregation. Biophys. J. 94:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brange, J., L. Andersen, E. D. Laursen, G. Meyn, and E. Rasmussen. 1997. Toward understanding insulin fibrillation. J. Pharm. Sci. 86:517–525. [DOI] [PubMed] [Google Scholar]
- 18.Jimenez, J. L., E. Nettleton, M. Bouchard, C. V. Robinson, C. Dobson, and H. Saibil. 2002. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA. 99:9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohren, C. F., and D. R. Huffman. 1983. Absorption and Scattering of Light from Small Particles. Wiley, New York.
- 20.Nielsen, L., S. Frokjaer, J. F. Carpenter, and J. Brange. 2001. Studies of the structure of insulin fibrils by Fourier transform infrared (FTIR) spectroscopy and electron microscopy. J. Pharm. Sci. 90:29–37. [DOI] [PubMed] [Google Scholar]
- 21.Bouchard, M., J. Zurdo, E. J. Nettleton, C. M. Dobson, and C. V. Robinson. 2000. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9:1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krebs, M. R. H., E. H. C. Bromley, and A. M. Donald. 2005. The binding of thioflavin-T to amyloid fibrils: localization and implications. J. Struct. Biol. 149:30–37. [DOI] [PubMed] [Google Scholar]
- 23.Sharp, J. S., J. A. Forrest, and R. A. L. Jones. 2002. Surface denaturation and amyloid fibril formation of insulin at model lipid-water interfaces. Biochemistry. 41:15810–15819. [DOI] [PubMed] [Google Scholar]