

Abstract
Summary
Several rules-based algorithms have been developed to interpret results of HIV-1 genotypic resistance tests. To assess the concordance of these algorithms and to identify sequences causing interalgorithm discordances, we applied four publicly available algorithms to the sequences of isolates from 2,045 individuals in northern California. Drug resistance interpretations were classified as S for susceptible, I for intermediate, and R for resistant. Of 30,675 interpretations (2,045 sequences × 15 drugs), 4.4% were completely discordant, with at least one algorithm assigning an S and another an R; 29.2% were partially discordant, with at least one algorithm assigning an S and another an I, or at least one algorithm assigning an I and another an R; and 66.4% displayed complete concordance, with all four algorithms assigning the same interpretation. Discordances between nucleoside reverse transcriptase inhibitor interpretations usually resulted from several simple, frequently occurring mutational patterns. Discordances between protease inhibitor interpretations resulted from a larger number of more complex mutation patterns. Discordances between nonnucleoside reverse transcriptase inhibitor interpretations were uncommon and resulted from a small number of individual drug resistance mutations. Determining the clinical significance of these mutation patterns responsible for interalgorithm discordances will improve interalgorithm concordance and the accuracy of genotypic resistance interpretation.
Keywords: HIV-1, Drug resistance, Genotype, Mutation, Reverse transcriptase, Protease
Several expert panels have recommended that drug resistance testing be used to help select optimal drug therapy for HIV-1—infected persons, and genotypic drug resistance testing has become routine in many developed countries (1,2). The manual interpretation of HIV-1 genotypic assays, however, is difficult because a large number of protease and reverse transcriptase (RT) drug resistance mutations interact and emerge in complex patterns. Several genotypic resistance interpretation algorithms have been developed that use protease and RT sequences to assess HIV-1 susceptibility to each of the available antiretroviral drugs. To quantify the extent of interalgorithm concordance and to identify mutations responsible for interalgorithm discordances, we applied four publicly available algorithms to a large set of clinical sequences. Here, we describe the results of these comparisons and the mutation patterns responsible for interalgorithm discordances.
MATERIALS AND METHODS
Sequences
Between July 1997 and December 2000, the Stanford University Hospital Diagnostic Virology Laboratory sequenced the protease and RT genes of HIV-1 isolates from ≈2,250 persons in northern California. This study used sequences from 2,045 persons that encompassed positions 1–99 of protease and 1–250 of RT; 205 sequences that did not encompass this entire region were excluded from our analysis. Sequencing was done as previously described (3). RNA was extracted from 0.2 mL of plasma using a guanidine-thiocyanate lysis reagent, reverse-strand cDNA was generated from viral RNA, and first-round PCR was done with Superscript One-Step RT-PCR (Life Technologies). Direct PCR cycle sequencing was performed using AmpliTaq DNA fluorescent sequencing polymerase and dRhodamine terminators (Applied Biosystems), and electropherograms were generated using an Applied Biosystems Model 377 sequencer. Each of the sequences has previously been submitted to GenBank (4).
Algorithms
Four publicly available, commonly used algorithms were applied to each sequence: Agence Nationale de Recherches sur le SIDA (ANRS) (5), updated March 2002 (ANRS-3–02); HIV RT and Protease Sequence Database (HIVDB) (http://hivdb.stanford.edu) (6), updated August 2002 (HIVDB-8–02); Rega Institute version 5.5 (Rega-5.5) (7); and Visible Genetics version 6 (Toronto, Ontario, Canada) (VGI-6) (8). Each algorithm accepts as input a list of mutations and generates output that categorizes anti-HIV drugs by the extent of their predicted activity.
ANRS-3–02, Rega-5.5, and VGI-6 are rules-based algorithms that each report three levels of resistance: susceptible (S), resistant (R), and an intermediate level, which is described differently for each algorithm but which we refer to here as intermediate (I). The HIVDB-8–02 algorithm assigns a drug penalty score for each drug resistance mutation. The total score for a drug is derived by adding the scores associated with each mutation. The program uses the total drug score to assign one of the following levels of inferred drug resistance: susceptible, potential low-level resistance, low-level resistance, intermediate resistance, and high-level resistance. For this study, “susceptible” and “potential low-level resistance” were considered susceptible (S), “low-level resistance” and “intermediate resistance” were considered intermediate (I), and “high-level resistance” was considered resistant (R).
Fifteen of the 16 U.S. Food and Drug Administration-approved antiretroviral drugs were studied. Zalcitabine was not included in the analysis, because it is not commonly used and few clinical data are available on zalcitabine resistance.
Mutations
Drug resistance mutations were defined as mutations associated with drug resistance in at least one of the rules-based algorithms. Mutations associated with nucleoside RT inhibitor (NRTI) resistance included M41L, E44AD, A62V, K65R, D67N, T69DGN, T69 insertions, K70R, L74V, V75IAMST, Y115F, F116Y, V118I, Q151M, M184VI, L210W, T215FY, and K219QNE. Mutations associated with nonnucleoside RT inhibitor (NNRTI) resistance included A98G, L100I, K101EQ, K103N, V106AI, V108I, V179DE, Y181CI, Y188LCH, G190ASET, P225H, M230L, and P236L. Mutations associated with protease inhibitor (PI) resistance included L10IFVR, K20RM, L24I, D30N, V32I, L33F, M36I, M46IL, I47V, G48V, I50V, F53L, I54AVLM, L63P, A71ILVT, G73S, V77I, V82ATFS, I84V, N88DS, L90M, and I93L.
The term “thymidine analog mutations” was used to refer to the common RT mutations: M41L, D67N, K70R, L210W, T215FY, and K219QE. The term “major protease mutations” was used to refer to protease mutations that alone were sufficient to influence the interpretation of at least one rule in at least one algorithm. These mutations included those at positions 30, 32, 46, 48, 50, 54, 82, 84, 88, and 90. The term “minor protease mutations” was used to refer to protease mutations that influenced the interpretation of one of the rules-based algorithms only in combination with other protease mutations.
Analysis
Each of the interpretation algorithms was implemented using the Algorithm Specification Interface on the Stanford HIV RT and Protease Sequence Database web site (http://hivdb.stanford.edu). Results obtained in this manner were independently verified for VGI-6 and ANRS-3–02 using software developed by Visible Genetics and for Rega-5.5 using software developed by the Rega Institute. Results were printed to a file containing 30,675 rows (2,045 sequences × 15 drugs) and individual columns for each interpretation and for each of the drug resistance mutations. The results file is available at http://hivdb.stanford.edu.
Interpretations were considered to be concordant if each of the four algorithms assigned the same level of resistance (S, I, or R) to a sequence for a particular drug. Partial discordances occurred when the algorithm’s interpretations were split between an S and an I or an I and an R. If at least one algorithm assigned an S and another assigned an R to the same sequence for a particular drug, then interpretations were said to be discordant.
Interalgorithm agreement was also quantified using the weighted κ statistic, which provides an indicator of general agreement. The four algorithms were grouped into six pairs. An agreement matrix was formed for each pairing using the assignment of S, I, or R for each sequence and drug, and then it was used to compute the weighted κ values. The analysis was done for each drug individually and for all of the drugs pooled.
The results file was processed by a program to produce the following output: the number of S, I, and R concordances and the number of S—I discordances, I—R discordances, and S—R discordances; a categorization of each S—R discordance by drug and pattern of algorithm assignments (i.e., because the algorithms were listed alphabetically, the pattern SIRI would indicate that ANRS-3–02 assigned an S, HIVDB-8– 02, an I, Rega-5.5, an R, and VGI-6, an I); and for the S—R discordances, a list of the frequency of each mutation and mutation combination within a set of sequences having the same pattern of algorithm assignments for a drug.
Identification of Mutation Patterns Responsible for Discordances
The theoretical upper limit for the number of different possible patterns of drug resistance mutations in a sequence, also known as a power set, is 2n, where n is the number of different drug resistance mutations. Because this number is very large—particularly if different amino acid substitutions at a single position are considered distinct mutations—two simplifications were introduced to generate lists of mutation patterns associated with S—R discordances having identical patterns of algorithm interpretations. These simplifications had no effect on how the interpretations were originally assigned.
First, mutations were divided into three groups: those present in at least one NRTI rule (NRTI resistance mutations), those in at least one NNRTI rule (NNRTI resistance mutations), and those in at least one PI rule (PI resistance mutations). Mutation patterns were then defined separately for each of these groups. Second, different substitutions at each drug resistance position were pooled, provided that the substitution was included in at least one rule.
The lists of mutation patterns and their frequencies within the sets of sequences associated with each pattern of interalgorithm discordances were examined to identify frequently occurring patterns responsible for the observed discordance. Many mutation patterns on the list were not responsible for the discordance; they simply represented a subset of mutations present within the set of sequences under analysis. Therefore, each pattern was submitted to the Algorithm Specification Interface to identify those patterns causing the observed interalgorithm discordances. Mutation patterns were considered to be easily identifiable when they were present in a large proportion of sequences.
RESULTS
Interalgorithm Discordances
Each algorithm assigned 30,675 drug resistance interpretations to 2,045 sequences. Complete concordance was found for 66.4% of interpretations: 47.5% were assigned an S by each algorithm; 18.3%, an R; and 0.6%, an I. Partial discordance was found for 29.2% of interpretations: 15.4% displayed S—I discordance, and 13.8% displayed I—R discordance. S—R discordances were found for 1,356 interpretations (4.4%). Lamivudine and the NNRTIs had the highest level of concordance (Figure 1). The NRTIs abacavir, didanosine, stavudine, and tenofovir and the PIs amprenavir and lopinavir had the lowest concordance (Figure 1).
FIG. 1.
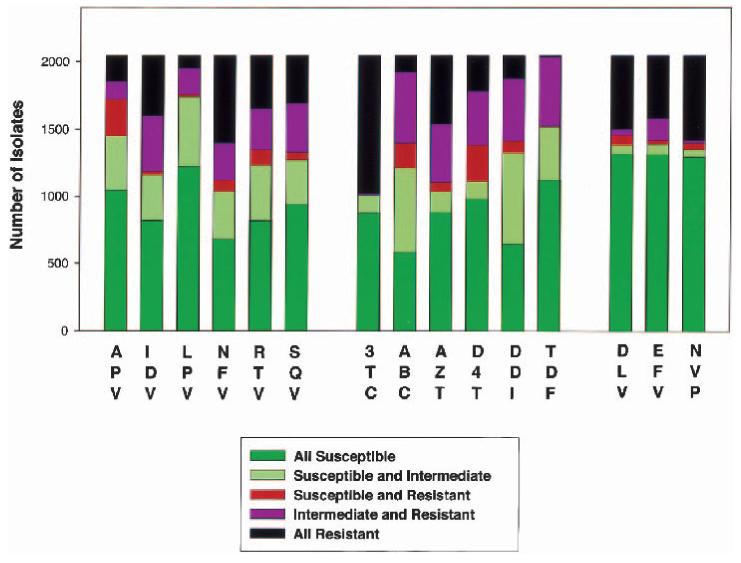
Nature of discordances between four HIV genotypic resistance interpretation algorithms according to drug. The results of 30,675 interpretations (2,045 sequences × 15 drugs) are summarized. Sequences for which all algorithms assigned an S (susceptible) are shown in green; those for which at least one algorithm assigned an S and another an I (intermediate) are shown in pale green; those for which at least one algorithm assigned an S and another an R (resistant) are shown in red; those for which at least one algorithm assigned an I and another an R are shown in purple; and those for which all algorithms assigned an R (or rarely an I) are shown in black.
Although an S—R discordance was present for 4.4% of interpretations, the level of S—R discordance between any two algorithms (as opposed to within the complete group of four algorithms) was only 1.13%, and it ranged from 0.03% between HIVDB-8–02 and VGI-6 to 2.8% between ANRS-3–02 and Rega-5.5. S—R discordances were inversely related to the frequency with which algorithms assigned an I rather than an S or R. HIVDB-8–02 assigned I to 25.1% of interpretations, Rega-5.5 assigned I to 18.4%, VGI-6 assigned I to 12.6%, and ANRS-3–02 assigned I to 4.0%. Because the level of concordance can be overly influenced by the frequent assignment of an I as opposed to an S or R, we also computed weighted κ values, which take into account S—I and I—R discordances as well as S—R discordances (Table 1).
TABLE 1.
Correlations (weighted kappa values) between genotype interpretation algorithms according to drug
| All | 3TC | ABC | AZT | d4T | ddI | TDF | DLV | EFV | NVP | APV | IDV | LPV | NFV | RTV | SQV | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANRS vs. HIVDB | 0.71 | 0.97 | 0.33 | 0.71 | 0.59 | 0.42 | 0.46 | 0.91 | 0.90 | 0.96 | 0.48 | 0.76 | 0.40 | 0.79 | 0.80 | 0.77 |
| ANRS vs. Rega | 0.70 | 0.93 | 0.32 | 0.84 | 0.59 | 0.35 | 0.51 | 0.90 | 0.89 | 0.93 | 0.49 | 0.80 | 0.67 | 0.69 | 0.59 | 0.73 |
| ANRS vs. VGI | 0.81 | 0.98 | 0.44 | 0.92 | 0.83 | 0.65 | 0.70 | 0.92 | 0.85 | 0.95 | 0.61 | 0.90 | 0.45 | 0.89 | 0.83 | 0.96 |
| HIVDB vs. Rega | 0.78 | 0.95 | 0.78 | 0.79 | 0.67 | 0.79 | 0.79 | 0.90 | 0.93 | 0.93 | 0.72 | 0.61 | 0.53 | 0.74 | 0.60 | 0.63 |
| HIVDB vs. VGI | 0.82 | 0.99 | 0.67 | 0.72 | 0.73 | 0.72 | 0.61 | 0.92 | 0.92 | 0.95 | 0.71 | 0.85 | 0.68 | 0.88 | 0.84 | 0.82 |
| Rega vs. VGI | 0.80 | 0.95 | 0.64 | 0.86 | 0.74 | 0.68 | 0.59 | 0.96 | 0.95 | 0.97 | 0.74 | 0.73 | 0.73 | 0.79 | 0.73 | 0.77 |
| Total | 0.77 | 0.96 | 0.53 | 0.81 | 0.69 | 0.60 | 0.61 | 0.92 | 0.91 | 0.95 | 0.63 | 0.77 | 0.57 | 0.80 | 0.73 | 0.78 |
3TC, lamivudine; ABC, abacavir; AZT, zidovudine; d4T, stavudine; ddI, didanosine; TDF, tenofovir; DLV, delavirdine; EFV, efavirenz; NVP, nevirapine; ANRS, ANRS-3-02; HIVDB, HIVDB-8-02; Rega, Rega-V5.5; VGI, VGI ver 6.0.
Nine hundred fifty-four (3.1%) of the HIVDB-8–02 interpretations were originally scored as having “potential low-level resistance.” Because these were considered S by our analysis, they contributed to 54 (4.0%) of the 1,356 S—R discordances.
Mutation Patterns Causing NRTI Discordances
Table 2 summarizes the mutation patterns causing NRTI S—R discordances. Eighty-seven percent (544/633) of these discordances resulted from abacavir, didanosine, or stavudine drug resistance interpretations. Although the overall concordance for tenofovir was low (Table 1 and Figure 1), there were few S—R discordances for this drug (most were S—I or I—R).
TABLE 2.
Mutation patterns causing discordances for nucleoside RT inhibitor genotypic resistance interpretations
| Interpretation |
|||||||
|---|---|---|---|---|---|---|---|
| Drug | No. discordances | A | H | R | V | No. sequences* | Mutation pattern† |
| 3TC | 3 | R | I | I | S | 3 | 67NG + 70R + 69ins(1AA)¶ + 219Q (3) |
| ABC | 184 | S | I | R | R | 103 | 41L + 184V + 215FY (87) |
| 41L + 74V + 215FY (7) | |||||||
| S | I | R | I | 40 | 184V + 215FY (38) | ||
| S | I | R | S | 19 | 74V + 184V (17) | ||
| 65R + 184V (2) | |||||||
| S | R | R | R | 10 | 74V + 184V + 215FY (4) | ||
| 41L + 74I + 184V + 215FY (3) | |||||||
| S | I | I | R | 6 | 41L + 69D + 215FY (4) | ||
| AZT | 70 | R | I | S | R | 36 | 184V + 215FY (36) |
| S | I | R | I | 28 | 2 non-215 TAMs (27) | ||
| R | S | S | R | 6 | 74IV + 184V + 215FY (5) | ||
| d4T | 273 | R | I | S | I | 217 | 215YF (217) |
| S | I | R | I | 41 | 67N + 69DN + 70R + 219QE (37) | ||
| R | I | S | R | 9 | 41L + 74V + 184V + 215FY (9) | ||
| R | S | S | I | 5 | 74V + 184V + 215FY (5) | ||
| ddI | 91 | R | I | S | I | 61 | 41L + 210W + 215Y (39) |
| 41L + 67N + 215FY (10) | |||||||
| 67N + 70R + 215FY (5) | |||||||
| S | I | I | R | 12 | 69D + 184V (9) | ||
| S | R | I | I | 7 | 74I + 184V (6) | ||
| S | I | S | R | 4 | 69D (4) | ||
| R | I | R | S | 3 | 67NG + 70R + 69ins(1AA)¶ + 219Q (3) | ||
| TDF | 12 | R | S | I | I | 9 | 41L + 67N + 184V + 215YF (8) |
| R | I | R | S | 3 | 67NG + 70R + 69ins(1AA)¶ + 219Q (3) | ||
3TC, lamivudine; ABC, abacavir; AZT, zidovudine; d4T, stavudine; ddI, didanosine; TDF, tenofovir; A, ANRS-3-02; H, HIVDB-8-02; R, Rega-V5.5; V, VGI ver 6.0; DRM, non-215 TAM, thymidine analog mutation other than one at position 215, M41L, D67N, K70R, L210W, K219QE.
Mutation patterns responsible for uncommon patterns of discordance are not shown. In addition to those listed, abacavir was responsible for six discordances (SRRI-3, SRII-2, SISR-1) and didanosine for eight discordances (SRIR-2, RRSI-1, SRIS-1).
The number of sequences with well-defined mutation patterns (last column) often does not add up to the total number of sequences because only those sequence containing common mutation patterns are listed. Listed patterns are non-overlapping: sequences matching two patterns were counted only once.
The single amino acid mutations at position 69 were T69T_L, T69T_T, and T69S_N, where the first amino acid is a substitution and the second is an insertion.
Ninety-one percent (578/633) of NRTI S—R discordances resulted from 21 easily identifiable, frequently occurring mutational patterns (Table 2). For example, T215YF alone was responsible for 217 stavudine discordances. The combination of M41L+M184V+T215YF was responsible for 87 abacavir discordances. The combination of M184V+T215YF was responsible for 38 abacavir and 36 zidovudine discordances. Other drug resistance mutations, including K65R, T69DN, L74VI, and other non-215 thymidine analog mutations, contributed to most of the remaining discordances. Of the 578 discordances caused by one of the 21 listed mutation patterns, 540 (93.4%) included at least one thymidine analog mutation.
Mutation Patterns Causing NNRTI Discordances
Table 3 summarizes the mutation patterns causing NNRTI S—R discordances. These discordances were much less frequent than NRTI and PI discordances and usually resulted from different interpretations of individual mutations rather than mutation combinations. Ninety-two percent of S—R discordances were caused by the following mutations: K101E, V106AI, V108I, Y181I, and M230L.
TABLE 3.
Mutation patterns causing discordances for non-nucleoside RT inhibitor genotypic resistance interpretations
| Interpretation |
|||||||
|---|---|---|---|---|---|---|---|
| Drug | No. discordances |
A | H | R | V | No. sequences* |
Mutation patterns |
| DLV | 75 | R | S | I | I | 23 | 101E (23)† |
| S | S | R | I | 15 | 108I (15) | ||
| S | S | R | S | 14 | 106I (14) | ||
| S | R | R | R | 12 | 181I (12) | ||
| S | R | R | I | 8 | 230L (8) | ||
| EFV | 22 | S | I | R | I | 10 | 230L (6) |
| 106A (4) | |||||||
| R | S | I | I | 9 | 101E (8)† | ||
| NVP | 49 | S | S | R | I | 15 | 108I (15) |
| S | S | R | S | 14 | 106I (14) | ||
| R | S | I | I | 8 | 101E (8)† | ||
| S | R | R | I | 8 | 230L (8) | ||
DLV, delavirdine; EFV, efavirenz; NVP, nevirapine; A, ANRS-3-02; H, HIVDB-8-02; R, Rega-V5.5; V, VGI ver 6.0.
Mutation patterns responsible for uncommon patterns of discordance are not shown. In addition to those listed, delavirdine was responsible for three discordances (SIRI-3), efavirenz for four discordances (SRRI-2, RRSR-1, SRSS-1), and nevirapine for four discordances (SIRI-3, SRSS-1).
The number of sequences with well-defined mutation patterns (last column) often does not add up to the total number of sequences because only those sequence containing common mutation patterns are listed. Listed patterns are nonoverlapping: sequences matching two patterns were counted only once. K101E caused fewer discordances for nevirapine and efavirenz than for delavirdine because this mutation was frequently accompanied by G190A, which causes resistance to nevirapine and efavirenz but not delavirdine.
Mutation Patterns Causing PI Discordances
Table 4 summarizes the mutation patterns causing PI S—R discordances. Amprenavir was responsible for 47% (272) of these 577 discordances. Although the overall concordance for lopinavir was low, there were few S—R discordances for this drug (most were S—I or I—R). The mutational patterns responsible for PI discordances were more complex and numerous than those causing NRTI discordances. Indeed, 28 different patterns were required to explain 74% (425/577) of S—R discordances. These 28 patterns included 18 that contained both major and minor mutations, 8 that contained only major mutations, and 4 that contained only minor mutations. Of the 425 discordances caused by one of the 28 listed mutation patterns, 288 (67.8%) required four or more mutations. In contrast, only 11.8% of the mutation patterns responsible for NRTI S—R discordances and none of those responsible for NNRTI S—R discordances required as many mutations.
TABLE 4.
Mutation patterns causing discordances for protease inhibitor genotypic resistance interpretations
| Interpretation |
|||||||
|---|---|---|---|---|---|---|---|
| Drug | No. discordant |
A | H | R | V | No. sequences* |
Mutation patterns† |
| APV | 272 | S | I | R | R | 86 | 46IL + 90M + 2 of (10IVFR, 20RMI, 63P) (46) |
| 82ATFS + 90M + 2 of (10IVFR, 20RMI, 63P) (15) | |||||||
| S | I | R | I | 61 | 54VTM + 82ATFS + 2 of (10IVFR, 20RMI, 63P) (26) | ||
| 46IL + 82ATFS + 2 of (10IVFR, 20RMI, 63P) (11) | |||||||
| S | I | I | R | 59 | 46IL + 90M (39) | ||
| 10IVFR + 48V + 54VTM + 82ATFS (6) | |||||||
| 82ATFS + 90M (5) | |||||||
| S | R | R | R | 33 | 84V + 90M + 2 of (10IVFR, 20RMI, 63P) (27) | ||
| S | R | I | R | 17 | 84V + 90M (10) | ||
| S | I | R | S | 11 | 10IVFR + 20RMI + 63P + 90M (8) | ||
| IDV | 27 | R | S | R | I | 10 | 46IL + 63P (10) |
| S | S | R | S | 7 | ≥4 minor mutations (7) | ||
| R | S | I | I | 4 | 46IL (4) | ||
| LPV | 18 | S | I | I | R | 18 | no apparent pattern |
| NFV | 83 | S | R | R | R | 34 | 46I + 82ATFS (19) |
| 48V + 82ATFS (11) | |||||||
| S | I | R | I | 16 | 82AFST + (63P ± 77I) (13) | ||
| S | I | R | R | 16 | 10IVFR + 54VTM + 82ATFS (10) | ||
| 46L + 82ATFS (4) | |||||||
| S | S | R | S | 7 | 36I + 63P + 71VT + 77I (7) | ||
| RTV | 117 | S | I | R | I | 74 | 46I + 88DS (10I or 63P) (28) |
| 90M + ≥3 minor mutations (18) | |||||||
| 46I + ≥3 minor mutations (18) | |||||||
| S | I | R | R | 18 | 46IL + 90M (12) | ||
| S | S | R | I | 11 | 46IL + (10I or 63P) (10) | ||
| S | S | R | S | 7 | ≥4 minor mutations (7) | ||
| SQV | 60 | S | I | R | I | 52 | 46IL + 82ATFS + 2 of (10IVFR + 63P + 71VTI) (32) |
| 10IVFR + 46IL + 54VT + 82ATFS (7) | |||||||
| S | I | R | S | 5 | 46IL + 4 minor mutations (5) | ||
APV, amprenavir; IDV, indinavir; LPV, lopinavir; NFV, nelfinavir; RTV, ritonavir; SQV, saquinavir; A, ANRS-3-02; H, HIVDB-8-02; R, Rega-V5.5; V, VGI ver 6.0.
Mutation patterns responsible for uncommon patterns of discordance are not shown. In addition to those listed, amprenavir was responsible for five discordances (SRRI-3, SRII-1, RIRS-1), indinavir for six discordances (SIRS-6), nelfinavir for 10 discordances (SIRS-4, RIIS-3, SIIR-2, SRRI-1), ritonavir for seven discordances (SIRS-6, SRRR-1), and saquinavir for three discordances (SSRS-3).
The definition of minor mutations is slightly different for each of the rules of each algorithm but generally includes several mutations from the following list: 10IVRF, 20RMI, 24I, 32I, 33VF, 36IV, 53L, 63P, 71VTI, 73CST, 77I, 88DST. The number of sequences with well-defined mutation patterns (last column) often does not add up to the total number of sequences because only those sequence containing common mutation patterns are listed. Listed patterns are non-overlapping: sequences matching two patterns were counted only once.
DISCUSSION
Genotypic resistance assays have become highly reproducible at determining the nucleotide sequence of HIV-1 RT and protease (3). However, because few clinicians are aware of the clinical significance of every drug resistance mutation, most genotypic assays use an interpretation algorithm to generate an inferred level of drug resistance based on mutations present in the sequence (9). The available HIV genotypic resistance interpretation algorithms have been developed by different experts or expert committees based on distillations of large amounts of published data on the phenotypic impact and clinical significance of drug resistance mutations. However, discordances between available drug resistance interpretation algorithms have been reported (10,11).
This study reports that most discordances among commonly used algorithms are partial: at least one algorithm assigned an S and another an I for the same sequence, or at least one algorithm assigned an I and another an R. Only 4.4% of sequences caused complete discordance, in which at least one algorithm assigned an S and another an R. Most discordances were attributed to drugs that require multiple mutations for the development of resistance. Drugs for which resistance usually emerges by the development of a single mutation—such as the NNRTIs and lamivudine—were responsible for few discordances.
Clinicians obtaining the results of a genotypic resistance assay usually need to know whether the genotype suggests that the patient will respond to a drug in a manner comparable with a patient with a wild-type isolate and, if not, whether the patient will obtain any antiviral benefit from a drug. For this reason, all four algorithms defined an intermediate level of resistance. However, the frequent assignment of intermediate levels of resistance by three of the four algorithms in this study, while reducing the number of S—R discordances, led to nearly 30% of sequences having an S—I or I—R discordance.
The primary purpose of this study was to identify mutational patterns responsible for interalgorithm discordances. A secondary purpose of this study was to establish the baseline level of concordance for future comparisons as existing algorithms are revised or new algorithms are developed. The study was not designed to compare the predictive value of algorithms because the subsequent treatment of and clinical outcome for the individuals from whom the sequenced isolates were obtained are not available.
This study has implications for clinicians and for experts developing algorithms for interpreting genotypic resistance assays. Clinicians faced with complicated genotypes should realize that not all genotypic interpretation algorithms provide the same resistance interpretation and should consider consulting with an expert before deciding on treatment for persons with complicated genotypic test results. The use of phenotypic tests in conjunction with genotypic tests may be particularly useful in this setting, provided the complementary nature of the information conveyed by each test is understood (12).
Experts developing algorithms should be aware that interalgorithm discordances can cause confusion among clinicians and patients and should routinely compare their interpretations with those produced by other algorithms. Mutation patterns associated with discordances should receive special attention when algorithms are updated. Finally, retrospective and prospective clinical studies should be performed to ascertain the clinical significance of the mutation patterns responsible for interalgorithm discordances.
Acknowledgments
The authors thank Luc Dehaspe and Elke Van Craenenbroeck (PharmaDM, Haasrode, Belgium) for help with the Rega-5.5 interpretation software and Matthew J. Gonzales (Stanford University, Stanford, CA) for help with data management. This work was supported in part by a Stanford University BioX Interdisciplinary Award and the AIDS Reference Laboratory of Leuven, which receives funding from the Belgian Ministry of Social Affairs through the Health Insurance System.
REFERENCES
- 1.Hirsch MS, Brun-Vezinet F, D’Aquila RT, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society—USA Panel. JAMA. 2000;283:2417–2426. doi: 10.1001/jama.283.18.2417. [DOI] [PubMed] [Google Scholar]
- 2.EuroGuidelines Group for HIV Resistance Clinical and laboratory guidelines for the use of HIV-1 drug resistance testing as part of treatment management: recommendations for the European setting. AIDS. 2001;15:309–320. doi: 10.1097/00002030-200102160-00003. [DOI] [PubMed] [Google Scholar]
- 3.Shafer RW, Hertogs K, Zolopa AR, et al. High degree of inter-laboratory reproducibility of human immunodeficiency virus type 1 protease and reverse transcriptase sequencing of plasma samples from heavily treated patients. J Clin Microbiol. 2001;39:1522–1529. doi: 10.1128/JCM.39.4.1522-1529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzales MJ, Machekano RN, Shafer RW. Human immunodeficiency virus type 1 reverse-transcriptase and protease subtypes: classification, amino acid mutation patterns, and prevalence in a northern California clinic-based population. J Infect Dis. 2001;184:998–1006. doi: 10.1086/323601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meynard JL, Vray M, Morand-Joubert L, et al. Phenotypic or genotypic resistance testing for choosing antiretroviral therapy after treatment failure: a randomized trial. AIDS. 2002;16:727–736. doi: 10.1097/00002030-200203290-00008. [DOI] [PubMed] [Google Scholar]
- 6.Shafer RW, Stevenson D, Chan B. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 1999;27:348–352. doi: 10.1093/nar/27.1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Laethem K, De Luca A, Antinori A, et al. A genotypic drug resistance interpretation algorithm that significantly predicts therapy response in HIV-1-infected patients. Antiviral Ther. 2002;7:123–129. [PubMed] [Google Scholar]
- 8.Reid C, Bassett R, Day S, et al. A dynamic rules-based interpretation system derived by an expert panel is predictive of virological failure. Antiviral Ther. 2002;7:S91. [Google Scholar]
- 9.Vandamme AM, Houyez F, Banhegyi D, et al. Laboratory guidelines for the practical use of HIV drug resistance tests in patient follow-up. Antiviral Ther. 2001;6:21–39. [PubMed] [Google Scholar]
- 10.Schmidt B, Walter H, Schwingel E, et al. Comparison of different interpretation systems for genotypic HIV-1 drug resistance data. Antiviral Ther. 2001;6:102. [Google Scholar]
- 11.Kijak GH, Rubio AE, Pampuro SE, et al. Discrepant results in the interpretation of HIV-1 drug-resistance genotypic data among widely used algorithms. HIV Med. 2003;4:72–78. doi: 10.1046/j.1468-1293.2003.00131.x. [DOI] [PubMed] [Google Scholar]
- 12.Parkin N, Chappey C, Maroldo L, et al. Phenotypic and genotypic HIV-1 drug resistance assays provide complementary information. J Acquir Immune Defic Syndr. 2002;31:128–136. doi: 10.1097/00126334-200210010-00002. [DOI] [PubMed] [Google Scholar]