

Abstract
Purpose
The purpose of this study was to investigate the expression and activity of protein tyrosine phosphatases (PTPs) in epithelium during corneal wound healing and how PTPs regulate activation of the c-Met receptor and their proximal signaling.
Methods
Rabbit corneas were injured by gently scraping the surface, leaving the limbal epithelium intact, and epithelium was collected at 1, 2, 3, and 7 days after the injury. In organ culture models, epithelium was removed and corneas were incubated with hepatocyte growth factor (HGF) with or without the PTP inhibitor bpV(phen), and the PI-3K inhibitors wortmannin and LY294002. Human corneal epithelial (HCE) cells were stimulated with HGF with or without bpV(phen). Total cell lysates and cytosolic and membrane fractions were analyzed by Western blot. PTP activities were measured with specific substrates. PTP1B and SHP-2 genes were knocked down by interference RNA (siRNA).
Results
PTP activity and expression increased during wound healing. The most abundant were SHP-2, PTP1B and PTEN. HGF activated the c-Met receptor in HCE cells up to 30 min and was downregulated by 2 hr. Inhibition of PTPs increased HGF-promoted wound healing, the HGF-activated phosphorylation of c-Met and its downstream signals PI-3K/Akt, but not ERK1/2 and p70S6K. PTP1B and SHP-2 were bound to the c-Met. Part of the c-Met was co-localized in the endoplasmic reticulum with PTP1B. PTP1B phosphorylation increased when the c-Met receptor was deactivated, and gene knockdown of PTP1B increased c-Met activation. SHP-2 phosphorylation and binding to c-Met was higher during receptor activation, and SHP-2 gene silencing decreased receptor phosphorylation.
Conclusions
Inhibition of PTPs activity mimics the effect of HGF by activating the PI-3K/Akt signal involved in wound healing. PTP1B and SHP-2 are bound to the c-Met receptor to control its activity. While binding of PTP1B increases when there is a decrease in c-Met activation and acts as a negative regulator of the receptor, increased binding and phosphorylation of SHP-2 coincide with maximal stimulation of c-Met, acting as a positive regulator.
INTRODUCTION
A central theme in corneal epithelial repair is how growth factors modulate the complex, highly interactive wound healing process (1–5). Regulation of cell proliferation, migration, adhesion and apoptosis is fundamental to obtaining an adequate repair of the epithelium and to maintaining corneal transparency. Growth factors exert their action through binding to receptor tyrosine kinases (RTK) that signal through lipid and protein kinases by specific phosphorylation-dephosphorylation reactions that will modulate the overall wound healing. RTKs contain an N-terminal extracellular binding protein, a transmembrane domain and a cytosolic C-terminal region with tyrosine kinase activity. In addition, many RTKs are coupled to a variety of adaptor proteins that enhance their responses (6). One of these RTKs is the c-Met receptor, whose ligand is hepatocyte growth factor (HGF). HGF is a paracrine growth factor that is released by corneal stroma cells and the lacrimal gland after cornea injury and acts on the c-Met in epithelial cells (7,8).
Our previous studies had shown that HGF activates a phosphatidylinositol-3 kinase (PI-3K)/Akt pathway involved in wound healing and survival (9,10) as well as the specific mitogen activated kinases, ERK1/2 and p38, which are important in epithelial cell proliferation and migration, respectively (11). Very recently, we have found that PKCα and PKCε are also activated by HGF and involved in the wound healing response of epithelial cells (12). Therefore, activity of c-Met must be tightly regulated in order to maintain normal cellular responses. Aberrant dysfunction of the receptor could be responsible for disorders in epithelial repair. In fact, during corneal wound healing, the activation of the PI-3K signaling is maintained for some time and then switched off, probably to avoid overactivation (13). One set of mechanisms that regulate cell signaling is protein tyrosine phosphatases (PTPs), which are enzymes that catalyze the de-phosphorylation of tyrosine phosphorylated proteins (14–17). PTPs can function as negative or positive regulators of signaling triggered by RTK. The PTPs comprise a very large family of phosphatases that are broadly classified into trans-membrane or receptor-like and non-trans-membrane or non-receptor PTPs. They are differentiated by their non-catalytic segments that are important for their cellular targeting. The nonreceptor PTPs are also structurally diverse. This allows them to target specific subcellular locations, including the cytosol, the plasma membrane, and the endoplasmic reticulum. They are also further divided according to their substrate specificity: tyrosine specific PTPs (such as PTP1B, PTP1C (also known as SHP-1), and PTP1D (also known as SHP-2)); dual specific phosphatases (DSPs), which have catalytic action in both phospho-Tyr and phospho-Ser or Thr (including enzymes such as mitogen-activated protein kinases (MAPK) phosphatases (MKPs), and Cdc25 (15)); and lastly the multiple-specific phosphatases (MSPs), which have additional substrate specificities (such as PTEN). The specificity of the PTPs relies also in the recognition of certain phosphopeptides for their action and in the selective expression of individual PTPs in cells and organellas.
There is not much information on PTPs in the cornea, except for a report on the expression of the trans-membrane PTP LAR in stroma cells of patients with keratoconus (18), a few more recent studies on the expression of some PTPs in endothelial cells that may be involved in maintaining the cells in a non-proliferative state (19,20), and about the association between changes in MKP-1 expression and migratory responses to EGF (21). Information on how PTPs are involved in the regulation of the c-Met receptor in the corneal epithelium is not available. In this study, we found that the corneal epithelium expresses a selective pattern of PTPs during wound healing. Inhibition of PTPs increases c-Met activation and cell signaling stimulated by HGF, as well as the rate of epithelial wound healing. Two non-receptor PTPs, PTP1B and SHP-2, increase their expression after epithelial injury and are bound to the c-Met receptor. While PTP1B is a negative regulator of c-Met activation, SHP-2 acts as an activator of the HGF receptor.
METHODS
Materials
Human recombinant double-chain HGF was a gift from Genentech (San Francisco, CA). The monoclonal antibodies PTP1B, SHP-1, SHP-2, LAR, and KAP, and the secondary antibodies against mouse and rabbit HRP conjugated were purchased from BD Biosciences Pharmingen (San Diego, CA). The mouse monoclonal antibodies c-Met, phosphotyrosine (PY) clone 4G10, the phosphorylated form of Akt (Ser 437), the rabbit polyclonal antibodies against the p85α subunit of PI3-kinase and the phosphorylated form of Met (Tyr 1234, Tyr 1235) were purchased from Upstate (Charlottesvile, VA). The rabbit polyclonal antibodies PTP1B, SHP-2, ERK1, the mouse monoclonal antibody of the phosphorylated form of p70S6K at Ser-411, the mouse monoclonal PTEN and GAPDH antibodies, the PTP1B substrate and the protein A or G agarose were from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit polyclonal antibody SHP-2 in its phosphorylated form at Tyr 542 and the biotinlyated protein marker detection kit were from Cell Signaling Technology (Beverly, MA). The mouse monoclonal active form of ERK1/2, sodium orthovanadate (SOV), and phenylarsine oxide (PAO) were purchased from Sigma-Aldrich (St. Louis, MO). The inhibitors bpV(phen), LY 294002 and wortmannin were from Calbiochem (La Jolla, CA). The CinnGEL 2Me inhibitor was purchased from Biomol (Plymouth Meeting, PA). Validated PTP1B siRNA, validated SHP-2 siRNA, negative siRNA #1, and siPORT Neo FX transfection reagent were purchased from Ambion (Austin, TX). The mouse monoclonal [RL90] to protein disulfide isomerase (PDI), an endoplasmic reticulum (ER) marker, was from Abcam (Cambridge, MA). The secondary antibodies used for immunostaining, goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 546, were from Invitrogen (Carlsbad, CA).
Corneal epithelial wound healing
I-“in vivo”model
New Zealand albino rabbits of both sexes weighing between 2–3 kg were used. The animals were treated in compliance with the guidelines of the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Ophthalmic and Vision Research, and the experimental protocol was approved by the Institutional Animal Care and Use Committee, Louisiana State University Health Sciences Center. The rabbits were anesthetized with a mixture of ketamine/xylazine injected intramuscularly. The corneal epithelium was gently scraped with a sterile blade, leaving the limbal epithelium intact and enabling the stem cells to differentiate, migrate and proliferate. This epithelium was used as control. Newly generated epithelial cells were removed at 1, 2, 3, and 7 days after injury by gently scraping the surface of the cornea and then homogenizing as described below. At the end of each time point, the rabbits were sacrificed with an overdose of pentobarbital.
II-Corneal organ culture wound-healing model
Rabbit eyes were obtained from Pel-Freez Biologicals (Roger, AR). The central part of the cornea was marked with a 7 mm surgical trephine and the epithelium was removed using a battery operated device (Algerbrush II, Alger Co., Lago Vista, TX) as previously described (11). The wounded corneas were excised with 1 mm rim and positioned in 6-well plate on sterile Teflon balls (12 mm diameter) (9). The wells were filled up to the cornea rim with DMEM/F12 medium with HGF (40ng/ml) with or without the PTP inhibitor bpV(phen) (6µM). In some wells the PI-3K inhibitors 200 nM wortmannin or 20 µM LY294002 were added. Corneas incubated without HGF and inhibitors were used as 24 h controls. To evaluate the wound healing, the corneas were stained with Alizarin red (9). Photographs were taken on a dissecting microscope with an attached camera and recorded by Adobe Photoshop software. The uncovered areas were analyzed by a computer digitizer and an image-analysis program.
PTP1B and SHP-2 siRNA transfections
HCE cells were transfected with PTP1B siRNA, SHP-2 siRNA, and negative control according to the manufacturer’s protocol. Briefly, for each well in a six well plate, 5 ul siPORT NeoFX transfection reagent was diluted with 100 :l Opti-MEM medium (Invitrogen) and incubated for 10 min at room temperature. Fifty pmoles of each siRNA (PTP1B, SHP-2, and negative control) were diluted in 100 :l of the same medium. Both solutions were mixed together and incubated for an additional 10 min at room temperature to ensure the formation of siRNA-transfection reagent complex, and then the mixture was dispensed into the well. HCE cells suspended in KGM (2.3 105/well) were overlaid on the transfection complexes. The plate was gently rocked back and forth to evenly distribute the cells into the complexes and incubated at 37°C for 24 h. The medium was replaced with fresh KGM and further incubated for 48 h. For the experiments, cells were starved overnight and either treated or not treated with 4 or 6 uM bpV(phen) for 30 min before being stimulated with 40 ng/ml HGF for 15 min or 2 h.
Cell culture
Human corneal epithelial (HCE) cells were used in this study. The HCE cell line was kindly provided by Roger Beuerman (LSU Eye Center, New Orleans, LA). The cell line was established using a HPV16-E6E7 vector (22), maintained as previously described (11), and used between passages 25–45. These cells responded to growth factors in a similar manner as the primary cultures (10–12, 23). When the cells reached 80–90 % confluence, they were switched to keratinocyte basal medium (KBM, Cambrex, Walkersville, MD) for 20 h. Cells were then stimulated with 40 ng/ml HGF. In some experiments, the PTP inhibitor bpV(phen) was added before HGF.
Cells fractionation
Cells collected from culture or from rabbit corneas wounded “in vivo” were homogenized using a glass-glass homogenizer in 20 mM Hepes pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM DTT, 10 µg/ml leupeptin, 25 µg/ml aprotinin, 1 mM PMSF, and 0.25 M sucrose and then centrifuged at 100,000 g for 1 h at 4°C. The supernatant, referred to as the cytosolic fraction, was collected and the pellet was resuspended in the same buffer containing 1 % Triton X-100, stirred for 1 h at 4°C and centrifuged as described above. This supernatant is referred to as the membrane fraction.
Immunoprecipitation and Western blot
HCE cells were lysated in 50 mM Tris HCl pH 7.6, 5 mM EGTA, 1 mM sodium orthovanadate, 1 % Triton X-100, 5 mM NaF, 1 mM sodium pyrophosphate, 5 mM β-glycero-phosphate, 150 mM NaCl, 2 mM DTT, 10 µg/ml leupeptin, 25 µg/ml aprotinin, 1 µM microcystin LR, and 1 mM PMSF (lysis buffer), and homogenized as explained before. The homogenate was centrifuged at 14,000 rpm for 15 min at 4°C and the pellet discarded.
HCE cell extracts containing 1 mg of protein were immunoprecipitated by incubating them with the antibodies for c-Met or phosphotyrosine (PY) for 2 h at 4°C. The immunocomplexes were captured with protein A or G agarose by incubating overnight. The agarose beads were collected by centrifugation and washed four times with lysis buffer. The immmunoprecipitates as well as cell extracts or cell fractions were resolved by SDS-PAGE, using pre-cast gels from Invitrogen (4–12 %), and transferred to PVDF membranes (Amersham, Piscataway, NJ). Biotinnylated protein molecular weight standards were applied in one line of each gel. The membranes were blocked with 5 % milk in Tris-buffered saline (TBS, 20 mM Tris-Cl, 150 mM NaCl, pH 7.6) plus 0.1 % Tween-20 for 1 h and then probed with specific primary antibodies, as described in Results, by incubating for 2 h at room temperature or overnight at 4°C. The membranes were washed with the same buffer and further incubated with appropriate HRP-conjugated secondary antibodies. To confirm similar loading, in appropriate experiments, the membranes were re-probed with control antibodies. The protein bands were visualized using chemilumuniscence detection reagents (ECL-Plus, Amersham) and quantified by densitometry.
Immunofluorescence staining
HCE cells were seeded in four-well glass slides, and upon reaching 50–60 % confluence, were starved for 24 h in KBM. The cells were stimulated with HGF (40 ng/ml) for 15 min. In some experiments, the cells were pre-treated for 30 min with 10 µM bpV(phen) before adding the growth factor. The cells were fixed with 2 % paraformaldehyde for 30 min, then blocked with 10 % normal goat serum, 1 % BSA in phosphate-buffered saline (PBS) for 30 min and incubated overnight at 4°C with the primary antibodies. Next, the cells were incubated with the appropriated secondary antibodies. After each step, the slides were washed three times with PBS. No staining was observed when the primary antibody was omitted. Hoechst staining was performed to localize the nuclei. Cells were examined with a Nikon Eclipse TE 200 fluorescence microscope and the images were captured with a Photometrics Cool Snap HQ camera.
Protein tyrosine phosphatase assay
In a 96-well microtiter plate (half area plate), 1–5 µg of protein from HCE cell fractions were incubated for 30 min at 30°C with the PTP substrate (RRLIEDAEpYAARG) or with the specific substrate for PTP1B (DADEpYIPQQG) in a final volume of 25 µl of assay buffer (25 mM HEPES pH 7.2, 50 mM NaCl, 5 mM DTT, and 2.5 mM EDTA). For PTP1B assays, inhibitors were added at the concentrations specified. The reaction was stopped by adding 100 µl Malachite green solution. After waiting 15 min for color development, activity was determined by reading the absorbency at 630 nm in a microtiter plate reader. Blanks were prepared without substrate. Reaction times and concentration of substrate and protein were tested in advance to ensure linearity of phosphate detection.
RESULTS
Changes in PTPs expression and activity during corneal epithelial wound healing in vivo
We first investigated whether there were changes in PTPs activity during corneal epithelial wound healing. Rabbit corneas were injured as explained in Methods, and tissue was collected 48 h after the wound; cytosolic and membrane fractions were isolated as described in Methods. The activity of the total PTPs was determined using a tyrosine phosphopeptide as substrate. There was a significant increase in the activity of PTPs after injury, with higher increases in the membrane fraction (Figure 1A). Next, we determined the expression of several PTPs by Western blot in cytosolic and membrane fraction of epithelium from rabbit corneas 1, 2, 3 and 7 days after injury (Figure 1B). In the cytosolic fraction, rabbit corneal epithelial cells expressed the non-receptor SHP-2 (also known as PTP1D or SH-PTP2) as well as PTEN, a MSP. Both PTPs increased their expression after injury. On the other hand, another non-receptor PTP, SHP-1 (also known as PTP1C or SH-PTP1) and a DSP called KAP had a very low rate of expression in the cytosolic fraction, but increased their expression 3 and 7 days after injury. Both of them were also expressed at different times after injury in the membrane fraction. The non-receptor PTP1B was the major PTP expressed in the membrane fraction, and its expression increased 1 day after injury and continued to increase (3.5 fold over control) by day 3. At 7 days, PTP1B expression was decreased to almost the same levels as controls. The epithelial membrane fraction also expressed LAR, a receptor type PTP, at 3 days and 7 days after injury. We found no expression for other PTPs tested (VHR, S1RPα1, RPTPβ, RPTPα).
Figure 1. PTP activity and expression during “in vivo” epithelial wound healing.
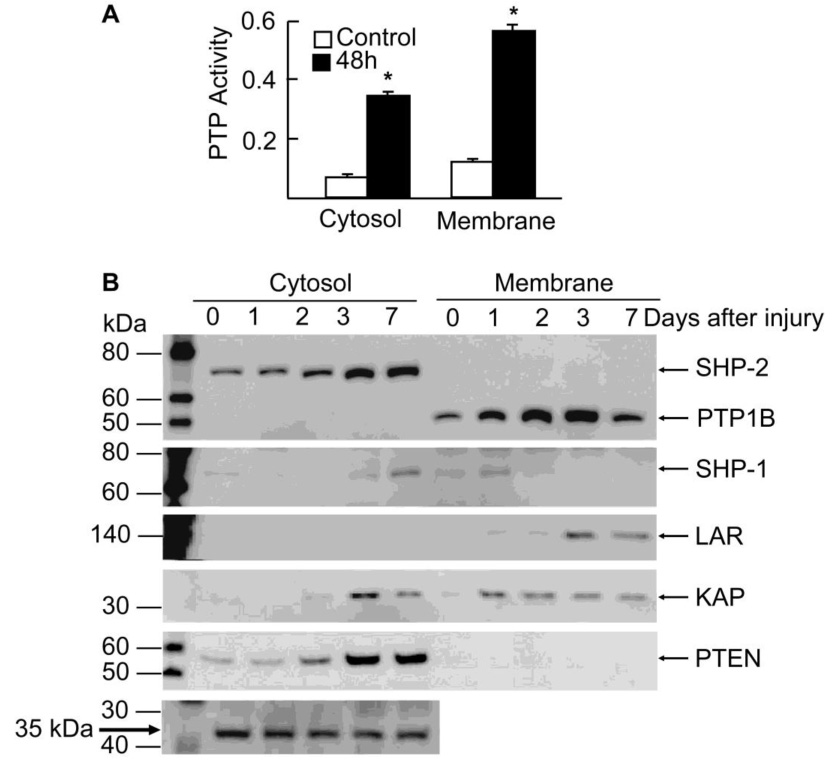
New Zealand rabbits were injured by gently scraping the corneas, leaving the limbal epithelium intact. Cornea epithelium from control and injured rabbits was collected and cytosol and membrane fractions prepared as explained in Methods. A) Total PTP activity was measured with 5 µg protein as explained in Methods before and after 48 h injury. Values represent the average ± SD of three samples. * significant differences with respect to control. B) Samples were collected 1, 2, 3 and 7 days after the injury and the same amount of proteins were separated by SDS polyacrilamide gel electrophoresis and analyzed by immunoblotting with PTPs antibodies. As loading controls for the cytosol fraction, a 35 kDa protein that appears in the gels after blotting with some of the antibodies, and did not change its expression during would healing, was used. In the membrane fraction, the phosphatase KAP also exhibited no change between 1–7 days after injury. The data represent one of two experiments.
PTPs inhibition increases c-Met phosphorylation in HCE cells
Activation of signal transduction pathways in response to HGF stimulation is mediated by autophosphorylation of two tyrosine residues, Tyr1234 and Tyr1235 localized in the intracellular region of c-Met (6). Using an antibody that recognizes these two phosphorylated residues, higher concentrations of HGF increased the tyrosine phosphorylation of c-Met, which appears as a specific band at 140 kDa in HCE cells (Figure 2A). In non-stimulated conditions, there was no visible tyrosine phosphorylation of c-Met. HGF at 40 ng/ml stimulated c-Met phosphorylation as soon as 5 min, continued to increase its activation up to 30 min, and decreased it by 60 min (Figure 2B). By 2 h, there was low activity, and none by 24 h. The results showed that in HCE cells, c-Met tyrosine phosphorylation stimulated by HGF was time- and concentration-dependent. Employing bpV(phen), a potent PTP inhibitor (24), we investigated the possibility of increasing the activation of c-Met by PTP inhibition. Cells were incubated in the presence of 1 and 10 µM bpV(phen) with or without HGF. Significant increased levels of p-c-Met were found after incubation with 10 µM bpV(phen) (Figure 2C). Stimulation with 40 ng/ml HGF for 15 min further enhanced the activation of c-Met. The blots were re-probed with GAPDH that showed no changes. Immunofluorescence also confirmed the presence of tyrosine phosphorylated c-Met after treatment with the PTP inhibitor, which was further increased in the presence of HGF (Figure 2D). The results demonstrate that PTP inhibition enhances HGF-induced activation of c-Met and suggest that bpV(phen) acts by preventing the de-phosphorylation and inactivation of c- Met through inhibition of associated PTPs.
Figure 2. Tyrosine phosphorylation of c-Met after HGF stimulation.
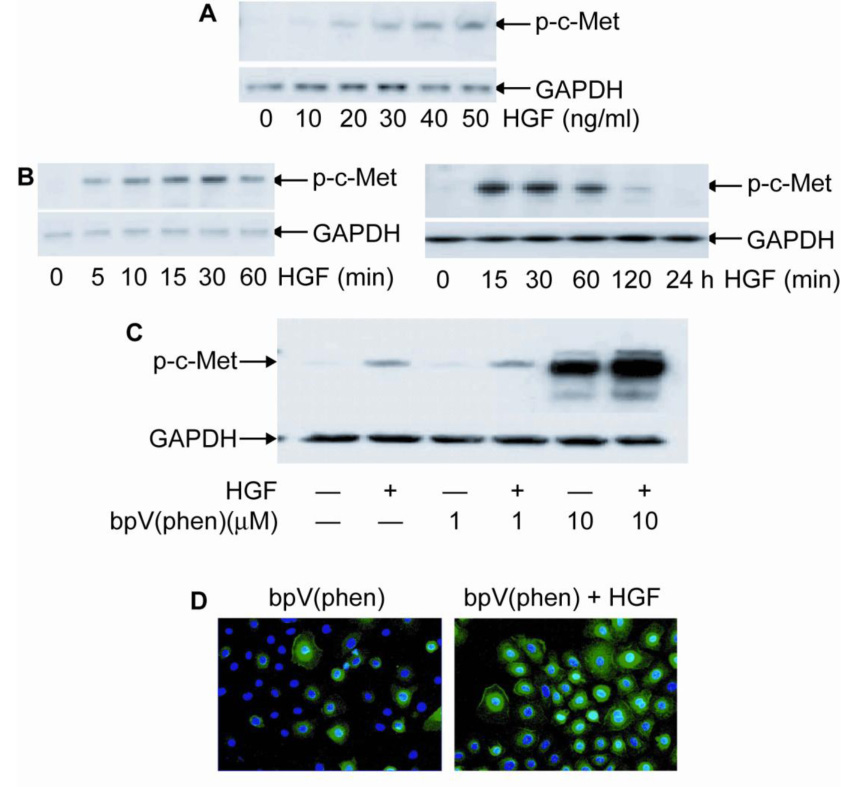
HCE cells were treated with HGF at concentrations and times indicated and then lysed. Whole cell lysates were immunoblotted with Tyr1234 and Tyr1235 phosphorylated c-Met antibody (p-c-Met). The membranes were re-blotted with GAPDH as gel loading control. In (A) HCE cells were incubated with different concentrations of HGF for 15 min, and in (B) and (C) stimulation was with 40 ng/ml HGF for different times. D) HCE cells were treated with bpV(phen) and stimulated or not with HGF for 15 min and immunostained with p-c-Met. Hoescht was use for nuclear counterstain. The experiments were repeated three times with similar results.
Inhibition of PTPs increase activation of PI-3K/Akt-1 pathway
We had shown previously that HGF stimulates PI-3K/Akt-1 signaling as well as p70S6K and ERK1/2 (9). Using HCE cells, we first studied the time-course activation of these kinases by HGF to determine if they follow similar patterns to that of c-Met phosphorylation. Phosphorylation of Akt-1 at Ser 437 was rapidly stimulated and elevation was maintained up to 1h (Figure 3A). This stimulation is similar to that reported in rabbit corneal epithelial cells (10). By 2 h, the activation was lower, and at 24 h it was similar to the control. Phosphorylation of p706SK and ERK1/2 followed the same profile as Akt-1. There were no changes in any of the total proteins with HGF. This demonstrates that tyrosine phosphorylation of c-Met is followed by activation of these kinases, and that when c-Met is down regulated, the kinases are also deactivated.
Figure 3. Effect of PTPs inhibition on HGF-stimulated downstream kinases.
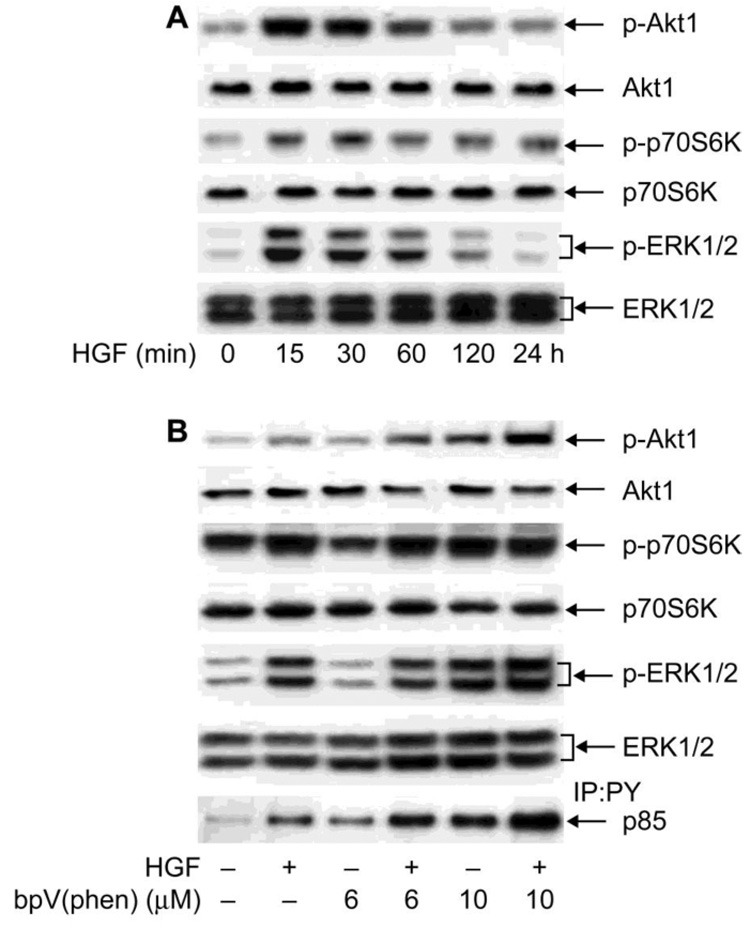
A) HCE cells were stimulated with 40 ng/ml HGF for different times and the cell lysate subjected to Western blot with: p-Akt-1, followed by re-blotting with Akt-1 antibody; p-p706SK followed by p706SK and p-ERK1/2 followed by ERK1/2 antibody. B) HCE cells were stimulated with HGF with or without 6 or 10 µM bpV (phen). To determine the phosphorylation of p85 subunit of PI-3K, the samples were first immunoprecipitated with PY antibody and then immunoblotted with p85. Data from one of three typical experiments are shown.
To investigate what role PTPs have in the signaling pathways activated by HGF, HCE cells were stimulated with 40 ng/ml HGF for 15 min with or without bpV(phen) (Figure 3B). As previously shown, 1 µM of the inhibitor did not cause an increase in c-Met phosphorylation (Figure 2C); therefore, for these experiments, 6 and 10 µM bpV(phen) were used. To determine activation of PI-3K, the cell extract was immunoprecipitated with phosphotyrosine (PY) and immunoblotted with the PI-3K regulatory subunit p85, which is bound to the c-Met complex and tyrosine phosphorylated during stimulation (6). HGF increased the amount of p85 present in anti-phosphotyrosine immunoprecipitates. The PTP inhibitor bpV(phen) also increased the phosphorylation of p85, which was further increased in cells stimulated with HGF. Similar patterns of activation were followed by Akt-1, a downstream target of PI-3K. However, phosphorylation of p706SK and ERK1/2 was increased only by the higher concentration of the inhibitor, 10µM, with no further increase in the presence of HGF. The results suggest that although inhibition of PTPs affects various signaling pathways, stimulation of PI-3K/Akt-1 linked to c-Met activation is more sensitive to regulation by PTPs.
PTP1B and SHP-2 complex with c-Met in HCE cells and are Tyrosine phosphorylated
HCE cells were stimulated at different times with HGF, and the cell extracts were immunoprecipitated with c-Met or PY antibodies as explained in Methods. Immunoblots with PTP1B showed a positive band at 50 kDa corresponding to the phosphatase (Figure 4A). Bound PTP1B to c-Met did not change with HGF stimulation up to 60 min, but continuous stimulation for 120 min and 24 h produced an increase that ranged from 35% to 60% of PTP1B bound to c-Met. The results correlate well with the data in Figure 2B, in which there was no tyrosine phosphorylation of c-Met at those times. The blots were reprobed with c-Met to demonstrate that the changes are not due to an increase in c-Met recovery from the immunoprecipitates. This demonstrates, for the first time, that PTP1B forms complexes with the c-Met receptor and that the bind increases after long-term stimulation with HGF.
Figure 4. Bound to c-Met and phosphotyrosine content of PTP1B and SHP2.
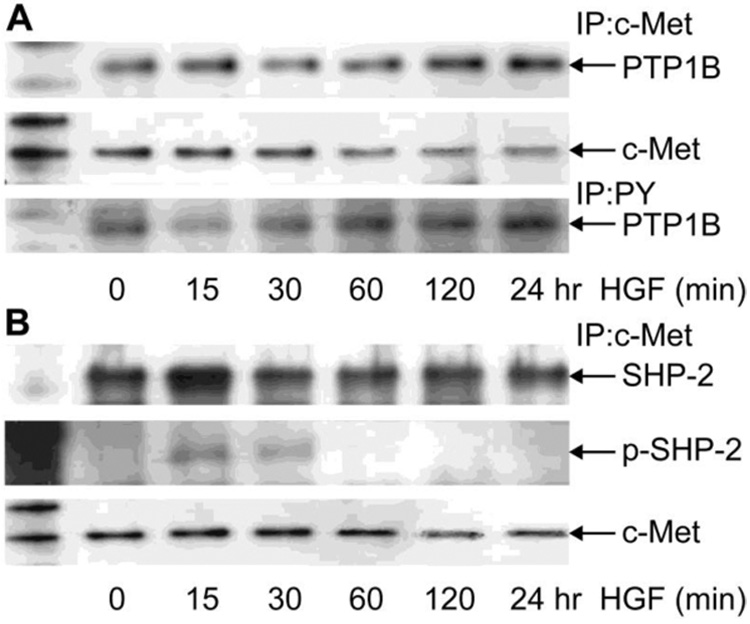
HCE cells were incubated in the absence or presence of 40 ng/ml HGF for different times and then collected in lysis buffer. Cell lysate (1mg) was immunoprecipitated with monoclonal c-Met antibody (A and B) or with PY monoclonal antibody (A) as indicated in Methods. Proteins were separated by 4–12 % gradient gels and transferred to PVDF membranes. The membranes were immunoblotted with the polyclonal PTP1B antibody (A) or with the polyclonal SHP-2 and the phosphorylated polyclonal p-SHP-2 antibody (B). The membranes were stripped and reprobed with c-Met antibody. The data represent one of three similar experiments.
PTP1B is a phosphoprotein whose activity can be regulated by phosphorylation at Ser/Thr or Tyr residues (25,26). To investigate the tyrosine phosphorylation state of PTP1B upon HGF stimulation, cell extracts from parallel experiments were immunoprecipitated with PY antibody and immunoblotted with PTP1B antibody (Figure 4A, lower panel). Non-stimulated HCE cells contain tyrosine phosphorylated PTP1B that decrease at 15 min and 30 min after HGF stimulation, but is elevated again at 60 min and remain higher up to 24 h. The increase in phosphotyrosine content in PTP1B correlates with the decrease in c-Met phosphorylation after HGF stimulation (Figure 2B), suggesting that tyrosine phosphorylation activates the phosphatase. It had been previously shown in other systems that the tyrosine phosphatase SHP-2 can be recruited to the c-Met and, upon stimulation, became tyrosine phosphorylated (6). This particular PTP, contrary to PTP1B, serves as a positive activator of the receptor for sustained activation of ERK1/2 (27). We investigated whether SHP-2 is bound and tyrosine phosphorylated after HGF stimulation in HCE cells. Immunoblots with SHP-2 antibody of c-Met immunoprecipitates from HCE cell extracts showed a strong band of SHP-2 (Figure 4B) that increased at 15 min stimulation. Bound SHP-2 was also phosphorylated at Tyr542 after 15 and 30 min stimulation. Immunoblotted controls with c-Met showed that the immunoprecipitates contain similar amounts of the receptor.
PTP1B and SHP-2 gene knock down affect the phosphorylation of c-Met
To obtain more clear evidence of the roles of PTP1B and SHP-2 on the phosphorylation of c-Met, the genes of these two PTPs were knocked down using validated siRNAs as described in Methods. Conditions were optimized for the maximum transfection efficiency. Ninety-six hours post–transfection, HCE cells were treated with HGF, 4 and 6 :M bpV(phen), or bpV(phen) plus HGF. These concentrations of inhibitor were chosen for the experiments in order to observe the changes in c-Met phosphorylation when the specific phosphatases were knocked down. The samples were analyzed by Western blot for protein expression using specific antibodies. HCE cells transfected with SHP-2 siRNA resulted in a 90 % decrease in SHP-2 expression compared to negative control siRNA (Figure 5A, upper bands). Cells transfected with PTP1B siRNA showed 70 % inhibition (Figure 5A, middle bands). GAPDH was used as a loading control. There was selectivity in the transfection, and HCE cells transfected with SHP-2 siRNA showed no changes in PTP1B expression compared to controls. Transfections with PTP1B siRNA showed only minimum changes of the expression of SHP-2. Figure 5B and 5C show how PTP1B and SHP-2-gene knockdown affected the phosphorylation of c-Met in the presence of bpV(phen). As already shown in Figure 2B, HGF at 15 min stimulates c-Met phosphorylation. Four :M bpV(phen) alone has no effect, but incubations in the presence of the inhibitor with 40 ng/ml HGF for 15 min increased the phosphorylation of c-Met, which was further increased (>80%) when PTP1B was knocked down. Decreased phosphorylation (20%) was observed when the cells were transfected with SHP-2 siRNA under the same conditions (Figure 5C). To further determine the role of PTP1B and SHP-2 in the activation of c-Met, HCE cells were incubated with 6 :M bpV(phen) for 2 h, a time that showed decrease in c-Met phosphorylation stimulated by HGF (Figure 2B). This concentration of the inhibitor stimulates the phosphorylation of c-Met at 2 h, which was further increased (>60 %) in presence of HGF. When PTP1B was knocked down (Figure 5B), a further 3-fold increase was observed, demonstrating that activation of PTP1B is important to switch off c-Met activation after HGF stimulation. However in the same conditions, phosphorylation of c-Met decreased (35%) when the cells were transfected with SHP-2 siRNA (Figure 5C), implicating that this phosphatase is a positive regulator of c-Met receptor in HCE cells.
Figure 5. Effect of PTP1B and SHP-2 knockdown on c-Met phosphorylation.
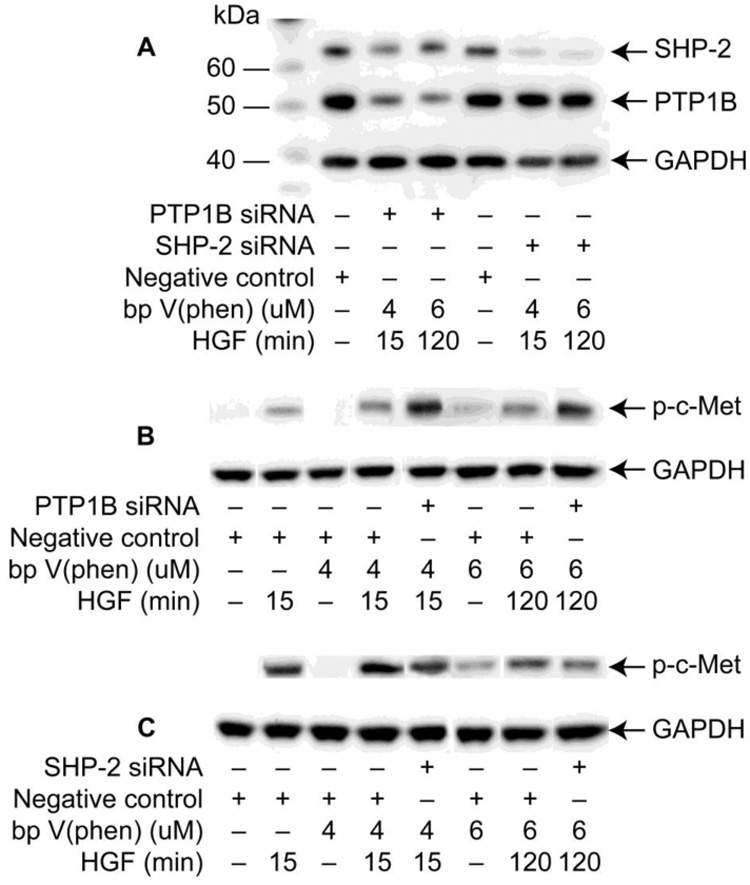
HCE cells were transfected with PTP1B, SHP-2, or negative control siRNA. Cells were treated with HGF, 4 or 6 M bpV(phen) or bpV(phen) plus HGF, as explained in Methods. (A) Selectivity of transfection and knockdown expression was assayed by immunobloting with PTP1B and SHP-2 antibodies. Cells transfected with PTP1B (B) or SHP-2 (C) and their controls were immunoblotted with p-c-Met antibody to show how the phosphorylation of c-Met is affected. GAPDH was used as loading control. The experiment was repeated once with similar results.
Cell localization of PTP1B and c-Met
The presence of PTP1B bound to c-Met in HCE cells prompted us to investigate the localization of these proteins in the cells. Immunofluorescence of HCE cells showed a strong PTP1B positive stain in the ER, which coincided with the PDI antibody (a marker of the ER) when the figures were merged (Figure 6A). No changes in PTP1B distribution were noted when the cells were stimulated with HGF (not shown). Interestingly, c-Met was also found around the nuclei with a considerable proportion of the growth factor receptor co-localized with PTP1B (Figure 6B).
Figure 6. Localization of PTP1B and c-Met in HCE cells.
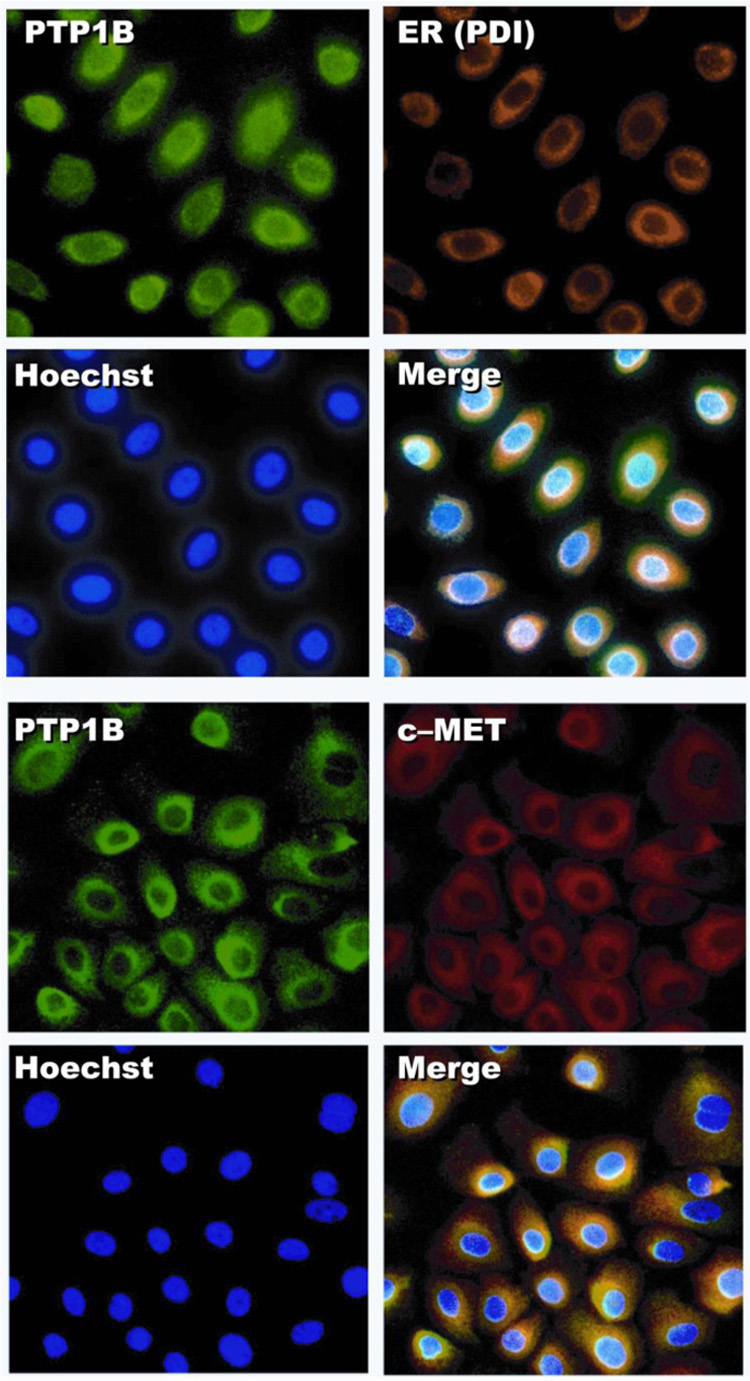
The cells were processed for immunofluoresence as described in Methods using PTP1B and c-Met antibodies. PDI (protein disulfide isomerase) antibody was used as an ER marker. Hoescht was employed to stain the nuclei. A) Co-localization of PTP1B with the ER marker is shown when the pictures were overlayed. B) Co- localization of PTP1B with c-Met.
Inhibition of PTP1B and increase in HGF-stimulated epithelial wound healing
Several vanadium derivative compounds had been shown to inhibit PTPs activity (28). In addition, derivatives of cinnamic acid had shown moderate selectivity for PTP1B inhibition (29). The action of four inhibitors at different concentrations on PTP1B activity was tested on the membrane fraction of HCE cells (Table 1). The most potent was bpV(phen), which at 6 µM produced 98 % inhibition of the PTP1B activity; PAO at 10 µM inhibited 56 % of the activity, while CinnGEL 2Me, considered a more specific PTP1B inhibitor (29), and SOV were much less potent. These results demonstrate that bpV(phen) at 6 µM is an excellent inhibitor of PTP1B in corneal epithelial cells. They also suggest that the increases in c-Met phosphorylation and PI-3K/Akt-1 signaling, after HGF stimulation in the presence of the inhibitor (Figure 2C and Figure 3B), could be due, in part, to the inhibition of this PTP.
Table I. Effect of tyrosine phosphatases inhibitors on PTP1B activity from membrane fraction of HCE cells.
HCE cells were incubated with the inhibitor at the concentrations indicated for 30 min (bpV(phen), PAO, or SOV) or 60 min (CinnGEL 2Me). One µg of protein from the membrane fraction was used to determine the PTP1B activity as explained in Methods. The percentage of inhibition was calculated with respect to control without inhibitors. Values correspond to average of 12 samples ± SD of three different experiments.
| Inhibitor | Concentration (µM) | % Inhibition |
|---|---|---|
| bpV(phen) | 1 | 12.3 ± 2.6 |
| 6 | 98.3 ± 0.4 | |
| 10 | 100.1 ±0.2 | |
| PAO | 1 | 25.6 ± 1.1 |
| 10 | 55.8 ± 1.8 | |
| CinnGEL Me 2Met | 1 | 12.6 ± 1.8 |
| 10 | 17.2 ± 2.7 | |
| 20 | 30.2 ± 3.8 | |
| SOV | 200 | 16.4 ± 3.0 |
| 1000 | 17.5 ± 3.5 |
We then investigated the action of bpV(phen) on epithelial wound healing. Rabbit corneas in organ culture were wounded as explained in Methods. As previously reported (9), HGF stimulated epithelial wound healing in this model 24 h after the wound (Figure 7). In the presence of 6 µM bpV(phen), there was a further decrease in the wounded area. The PI-3K inhibitors, wortmannin and LY294002, on the other hand, blocked epithelial wound healing stimulated by the PTP inhibitor. These experiments demonstrate that inhibition of PTP action increases HGF stimulation of corneal epithelial wound healing that requires activation of PI-3K signaling.
Figure 7. bpV(phen) increases HGF-stimulated epithelial wound healing that is reversed by PI-3K inhibitors.
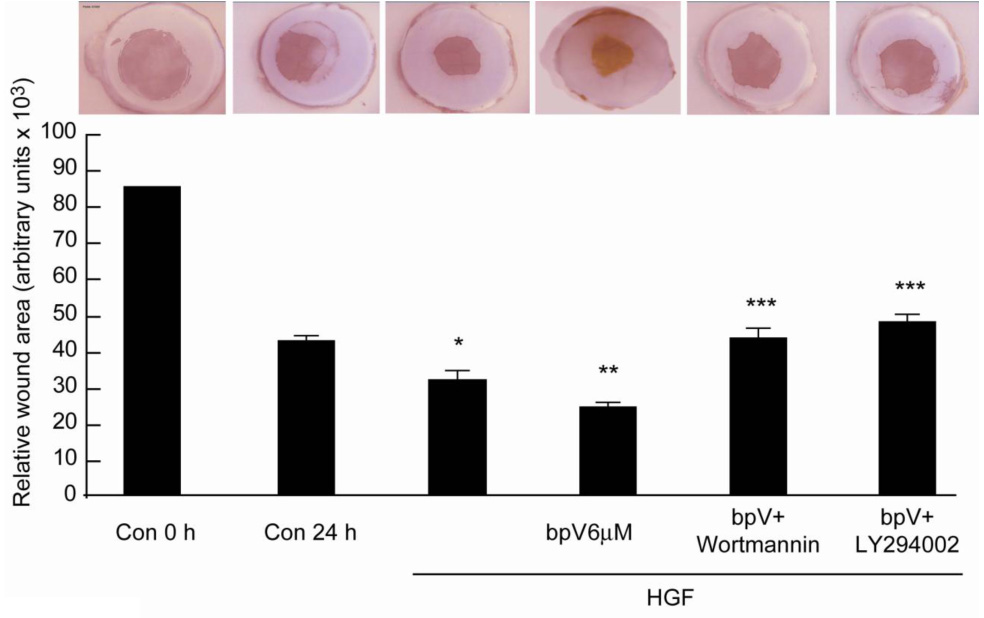
Corneas were injured and cultured in a serum free medium for 24 h in the presence of HGF (40 ng/ml) with or without 6 µM bpV( phen) or with or without 200 nM wortmannin or 20 µM LY294002. The corneas were stained with Alizarin red and the remaining uncovered area calculated. The pictures represent a cornea in each condition. The bars correspond to 5–6 corneas in each treatment. *p<0.05 compared to control at 24 h, **p< 0.05 compared to HGF, ***p< 0.05 compare to bpV(phen) + HGF (Student test analysis).
DISCUSSION
Corneal epithelial injury “in vivo” is a complex process that involves an array of growth factors and cytokines that activate a variety of cell signaling mechanisms to induce the repair of tissue. In this study, we found that after an epithelial wound, there was a significant increase in PTP activity in both the cytosol and membrane fraction of epithelial cells. Although the increase corresponded to total PTPs activity, PTP1B expression increased significantly 2 days after the injury and made for a proportion of the increase in activity. After in vivo corneal epithelial injury, PTPs were expressed in different compartments and at different rates. Some of them, such as SHP-2 and PTEN continued to increase their expression up to 7 days after injury, while others, such as PTP1B and KAP, had a peak at 3 days, suggesting that they regulate different cell signaling after injury. Therefore, selectivity at the level of PTPs is an important mechanism in regulating cell adhesion, migration, survival and proliferation of corneal epithelial cells. For example, KAP had been reported to be involved in the phosphorylation of cyclin-dependent kinases (30), and its increase in expression after injury could be linked to inactivation of the kinases and inhibition of cell proliferation. This could explain the appearance of KAP in the cytosol at longer times when the epithelium had been regenerated. This may be important in controlling mitosis at the level of CDKs (31). Another PTP that changed after injury was PTEN. This enzyme belongs to the MSP and is able to dephosphorylate protein tyrosine residues and the D-3 phosphate of inositol phospholipids. Although there is an increase of PTEN expression during the wound healing process, it is present in the cytosolic compartment, and does not bind directly to RTK receptors (13), suggesting that its regulation is at another level. LAR was expressed in epithelium at 3 and 7 days after injury. This enzyme corresponds to the receptor-like PTPs; it is known to bind to laminin and has been postulated to be involved in cell adhesion (32). Very recently, it has been shown that LAR dephosphorylates c-Met in confluent hepatocytes (33). This could suggest an important role in controlling the proliferation of the cells. It had also been reported to be a negative regulator of insulin signaling (34). Further studies are warranted to explore the functions of PTEN and LAR in corneal epithelial repair.
The non-receptor PTP1B and SHP-2 were highly expressed during wound healing, and we focus our attention on these two PTPs as candidates to modulate HGF signaling. PTP1B has been shown to regulate insulin and insulin-like growth factor-1 signaling in vivo and in vitro and has been implicated as a modulator of diabetes and obesity (26,35,36). It also regulates the action of EGF, platelet-derived growth factor and fibroblast growth factor (37,38), and there is some recent evidence that it can regulate EGF receptors in corneal epithelial cells (20). However, there is only one recent study to link this PTP with c-Met, in which it was reported that PTP1B-deficient hepatocytes show an increase in c-Met phosphorylation (39). To our knowledge, this is the first report to demonstrate association of the c-Met receptor with PTP1B. Evidence of the involvement of PTP1B in HGF action is provided by showing that binding to c-Met increased at longer times of HGF stimulation and correlates well with dephosphorylation of c-Met and down regulation of signaling pathways. In addition, knocking down the expression of PTP1B increased c-Met phosphorylation.
It is important to note that PTP1B bound to c-Met when there was no HGF stimulation. In experiments in kidney cells, interaction between insulin receptor and PTP1B occurs under basal conditions, and it was suggested that this interaction is important during insulin receptor biosynthesis in the ER to maintain the receptor in a non-activated form (40). In HCE cells, most of the PTP1B was localized in the ER. Therefore, this localization might be important for the regulation of c-Met receptors during their biosynthesis. In addition, to avoid overactivation of the receptor, c-Met could be internalized, but must be also de-phosphorylated to prevent stimulation of intracellular signaling pathways in an anomalous way. In fact, immunostaining shows that a significant proportion of c-Met co-localized with PTP1B. Interaction of this phosphatase with c-Met at the plasma membrane is a less possible scenario since we could not detect immunostaining in the cell membrane.
One way to regulate PTP1B is by phosphorylation. Several phosphorylation sites have been identified (25, 26). Although changes in activity had been associated with changes in its phosphorylation, the precise mechanisms by which the phosphorylation of PTP1B can be important in activating and inhibiting RTKs are unclear. Our results show that PTP1B is tyrosine phosphorylated. This phosphorylation decrease, at 15 and 30 min after HGF stimulation, coincides with maximal activation of c-Met. This suggests a positive action, by which phosphorylated PTP1B might increase its phosphatase activity. This in turn allows c-Met to be de-phosphorylated and inactivated at longer times. In fact, in knocking down PTP1B, there was further increase in c-Met phosphorylation stimulated by HGF in the presence of bpV(phen). The results are in agreement with previous studies showing that, in fibroblasts, insulin stimulates tyrosine phosphorylation of PTP1B and activates the enzyme (26).
Another important PTP in the regulation of the HGF receptor is SHP-2, which acts as a positive modulator of c-Met (27). This is also demonstrated in HCE cells, in which SHP-2-gene silencing decreased the activation of c-Met induced by HGF in the presence of the PTP inhibitor. The HGF receptor binds to several adaptor proteins (such as Grb1, ShC, Crk and Gab1) that, in turn, recruit other proteins involved in cell signaling. For example, the binding protein Gab1 recruits SHP-2 (6). This particular PTP is mainly cytosolic and increases its expression during wound healing. We had found a portion of the enzyme bound to the c-Met receptor in unstimulated cells that increased after 15 min of stimulation. That probably accounts for a very small proportion of the enzyme, which is difficult to detect in the membrane fraction without c-Met immunoprecipitation. In fact, our Western blots of epithelium collected during wound healing show a tenuous band in the membrane fraction.
In epithelial kidney cells, SHP-2 activation is required for tubulogenesis of the cells as well as for sustained activation of ERK1/2 (27). Our data on HCE cells show that ERK1/2 is down regulated by 2 h after HGF stimulation, while phosphorylation (activation) of SHP-2 lasts for 30 min upon HGF stimulation. This suggests that SHP-2 controls HGF responses during the early phase of c-Met activation. The transient activation of SHP-2 may be insufficient for modulating cell proliferation that is stimulated by ERK1/2 in corneal epithelial cells (11). It has also been suggested that SHP-2 could play a role in regulating cell spreading and migration (41). In that context, we had previously shown that during the epithelial migration phase, there is an inhibition of ERK1/2 activation (11). It is tempting to suggest that SHP-2 could be a regulator in that particular phase of epithelial wound healing. Future experiments will be needed to investigate this interesting point.
Finally, we also demonstrated that inhibiting PTPs activity with bpV(phen) increased HGF-stimulated epithelial wound healing, which was reverted when the PI-3K pathway was blocked. Time-response experiments with the PTPs inhibitor in the presence of HGF demonstrated that c-Met phosphorylation correlates well with activation of PI-3K/Akt signal pathway. Although these studies were done with a broad PTP inhibitor, we showed that bpV(phen) completely blocked PTP1B activity. This, along with our experiments with PTP1B siRNA, suggests that this particular phosphatase is a key modulator of corneal epithelial wound healing stimulated by HGF.
In summary, our data show that there is an increase in activity and expression of selective PTPs after corneal epithelial injury. Inactivation of PTPs selectively increases the PI-3K/Akt-1 pathway stimulated by HGF and the wound healing response of epithelium. SHP-2 binds to c-Met in HCE cells, its phosphorylation (activation) state coincides with the maximal c-Met activation, and gene knockdown of SHP-2 decreases the stimulated c-Met phosphorylation, which all demostrates a positive role of SHP-2 in the HGF action on HCE cells. We also demonstrate, for the first time, that PTP1B is bound to the c-Met receptor and that its tyrosine phosphorylation is down regulated when HGF increases the activity of c-Met and its downstream signals. PTP1B gene silencing increases the activation of c-Met, demonstrating an inhibitory role of this phosphatase. This new action of PTP1B on c-Met opens novel possibilities to explore the role of this particular phosphatase in regulating the actions of HGF, not only in corneal epithelium, but in other tissues in which c-Met has important functions.
Acknowledgements
This research was supported by United States Public Health Service grant R01 EY006635 from the National Eye Institute, National Institutes of Health, Bethesda, Maryland.
Supported by NIH EY006635
References
- 1.Schultz G, Khaw PT, Oxford K, MaCauley S, Van Setten G, Chegini N. Growth factors and ocular wound healing. Eye. 1994;8(Pt 2):184–187. doi: 10.1038/eye.1994.43. [DOI] [PubMed] [Google Scholar]
- 2.Imanishi J, Kamiyama K, Iguchi I, Kita M, Sotozono C, Kinoshita S. Growth factors: importance in wound healing and maintenance of transparency of the cornea. Prog Retin Eye Res. 2000;19:113–129. doi: 10.1016/s1350-9462(99)00007-5. [DOI] [PubMed] [Google Scholar]
- 3.Tervo T, van Setten GB, Paallysaho T, Tarkkanen A, Tervo K. Wound healing of the ocular surface. Ann Med. 1992;24:19–27. doi: 10.3109/07853899209164141. [DOI] [PubMed] [Google Scholar]
- 4.Gipson IK, Inatomi T. Extracellular matrix and growth factors in corneal wound healing. Curr Opin Ophthalmol. 1995;6:3–10. doi: 10.1097/00055735-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Lu L, Reinach PS, Kao WW. Corneal epithelial wound healing. Exp Biol Med (Maywood) 2001;226:653–664. doi: 10.1177/153537020222600711. [DOI] [PubMed] [Google Scholar]
- 6.Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–5589. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- 7.Wilson SE, Chen L, Mohan RR, Liang Q, Liu J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp Eye Res. 1999;68:377–397. doi: 10.1006/exer.1998.0603. [DOI] [PubMed] [Google Scholar]
- 8.Wilson SE, Liang Q, Kim WJ. Lacrimal gland HGF, KGF, and EGF mRNA levels increase after corneal epithelial wounding. Invest Ophthalmol Vis Sci. 1999;40:2185–2190. [PubMed] [Google Scholar]
- 9.Chandrasekher G, Kakazu AH, Bazan HE. HGF- and KGF-induced activation of PI-3K/p70 s6 kinase pathway in corneal epithelial cells: its relevance in wound healing. Exp Eye Res. 2001;73:191–202. doi: 10.1006/exer.2001.1026. [DOI] [PubMed] [Google Scholar]
- 10.Kakazu A, Chandrasekher G, Bazan HE. HGF protects corneal epithelial cells from apoptosis by the PI-3K/Akt-1/Bad- but not the ERK1/2-mediated signaling pathway. Invest Ophthalmol Vis Sci. 2004;45:3485–3492. doi: 10.1167/iovs.04-0372. [DOI] [PubMed] [Google Scholar]
- 11.Sharma GD, He J, Bazan HE. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem. 2003;278:21989–21997. doi: 10.1074/jbc.M302650200. [DOI] [PubMed] [Google Scholar]
- 12.Sharma GD, Kakazu A, Bazan HEP. Protein kinase C alpha and epsilon differentially modulate hepatocyte growth factor-induced proliferation and migration. 2006 doi: 10.1016/j.exer.2007.05.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandrasekher G, Bazan HE. Corneal epithelial wound healing increases the expression but not long lasting activation of the p85alpha subunit of phosphatidylinositol-3 kinase. Curr Eye Res. 1999;18:168–176. doi: 10.1076/ceyr.18.3.168.5372. [DOI] [PubMed] [Google Scholar]
- 14.Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- 15.Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 16.Ostman A, Bohmer FD. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001;11:258–266. doi: 10.1016/s0962-8924(01)01990-0. [DOI] [PubMed] [Google Scholar]
- 17.Gee CE, Mansuy IM. Protein phosphatases and their potential implications in neuroprotective processes. Cell Mol Life Sci. 2005;62:1120–1130. doi: 10.1007/s00018-005-5008-4. [DOI] [PubMed] [Google Scholar]
- 18.Chiplunkar S, Chamblis K, Chwa M, Rosenberg S, Kenney MC, Brown DJ. Enhanced expression of a transmembrane phosphotyrosine phosphatase (LAR) in keratoconus cultures and corneas. Exp Eye Res. 1999;68:283–293. doi: 10.1006/exer.1998.0604. [DOI] [PubMed] [Google Scholar]
- 19.Chen WL, Harris DL, Joyce NC. Effects of SOV-induced phosphatase inhibition and expression of protein tyrosine phosphatases in rat corneal endothelial cells. Exp Eye Res. 2005;81:570–580. doi: 10.1016/j.exer.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Harris DL, Joyce NC. Protein tyrosine phosphatase, PTP1B, expression and activity in rat corneal endothelial cells. Mol Vis. 2007;13:785–796. [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Yang H, Tachado SD, et al. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen DH, Bueurman RW, De Wever B, Rosdy M. Three-dimensional construct of the human corneal epithelium in vivo toxicology. In: Salem H, Katz SA, editors. Alternative Toxicology Methods. 2003. pp. 147–159. [Google Scholar]
- 23.Taheri F, Bazan HE. Platelet-activating factor overturns the transcriptional repressor disposition of Sp1 in the expression of MMP-9 in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2007;48:1931–1941. doi: 10.1167/iovs.06-1008. [DOI] [PubMed] [Google Scholar]
- 24.Band CJ, Posner BI, Dumas V, Contreres JO. Early signaling events triggered by peroxovanadium [bpV(phen)] are insulin receptor kinase (IRK)-dependent: specificity of inhibition of IRK-associated protein tyrosine phosphatase(s) by bpV(phen) Mol Endocrinol. 1997;11:1899–1910. doi: 10.1210/mend.11.13.0041. [DOI] [PubMed] [Google Scholar]
- 25.Ravichandran LV, Chen H, Li Y, Quon MJ. Phosphorylation of PTP1B at Ser(50) by Akt impairs its ability to dephosphorylate the insulin receptor. Mol Endocrinol. 2001;15:1768–1780. doi: 10.1210/mend.15.10.0711. [DOI] [PubMed] [Google Scholar]
- 26.Dadke S, Kusari A, Kusari J. Phosphorylation and activation of protein tyrosine phosphatase (PTP) 1B by insulin receptor. Mol Cell Biochem. 2001;221:147–154. doi: 10.1023/a:1010909031310. [DOI] [PubMed] [Google Scholar]
- 27.Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–8525. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang ZY. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 29.Moran EJ, Sarshar S, Cargill JF, et al. Radio frequency tag encoded combinatorial library method for discovery of tripeptide-substituted cinnamic acid inhibitors of the protein tyrosine phosphatase PTP1B. J Am Chem Soc. 1995;117:10787–10788. [Google Scholar]
- 30.Hannon GJ, Casso D, Beach D. KAP: a dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:1731–1735. doi: 10.1073/pnas.91.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zieske JD, Francesconi CM, Guo X. Cell cycle regulators at the ocular surface. Exp Eye Res. 2004;78:447–456. doi: 10.1016/s0014-4835(03)00205-7. [DOI] [PubMed] [Google Scholar]
- 32.O'Grady P, Thai TC, Saito H. The laminin-nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR. J Cell Biol. 1998;141:1675–1684. doi: 10.1083/jcb.141.7.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machide M, Hashigasako A, Matsumoto K, Nakamura T. Contact inhibition of hepatocyte growth regulated by functional association of the c-Met/hepatocyte growth factor receptor and LAR protein-tyrosine phosphatase. J Biol Chem. 2006;281:8765–8772. doi: 10.1074/jbc.M512298200. [DOI] [PubMed] [Google Scholar]
- 34.Mander A, Hodgkinson CP, Sale GJ. Knock-down of LAR protein tyrosine phosphatase induces insulin resistance. FEBS Lett. 2005;579:3024–3028. doi: 10.1016/j.febslet.2005.04.057. [DOI] [PubMed] [Google Scholar]
- 35.Cheung A, Kusari J, Jansen D, Bandyopadhyay D, Kusari A, Bryer-Ash M. Marked impairment of protein tyrosine phosphatase 1B activity in adipose tissue of obese subjects with and without type 2 diabetes mellitus. J Lab Clin Med. 1999;134:115–123. doi: 10.1016/s0022-2143(99)90115-4. [DOI] [PubMed] [Google Scholar]
- 36.Taghibiglou C, Rashid-Kolvear F, Van Iderstine SC, et al. Hepatic very low density lipoprotein-ApoB overproduction is associated with attenuated hepatic insulin signaling and overexpression of protein-tyrosine phosphatase 1B in a fructose-fed hamster model of insulin resistance. J Biol Chem. 2002;277:793–803. doi: 10.1074/jbc.M106737200. [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem J. 1997;327(Pt 1):139–145. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang Y, Ceacareanu B, Zhuang D, et al. Counter-regulatory function of protein tyrosine phosphatase 1B in platelet-derived growth factor- or fibroblast growth factor-induced motility and proliferation of cultured smooth muscle cells and in neointima formation. Arterioscler Thromb Vasc Biol. 2006;26:501–507. doi: 10.1161/01.ATV.0000201070.71787.b8. [DOI] [PubMed] [Google Scholar]
- 39.Sangwan V, Paliouras GN, Cheng A, Dube N, Tremblay ML, Park M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. J Biol Chem. 2006;281:221–228. doi: 10.1074/jbc.M507858200. [DOI] [PubMed] [Google Scholar]
- 40.Issad T, Boute N, Boubekeur S, Lacasa D. Interaction of PTPB with the insulin receptor precursor during its biosynthesis in the endoplasmic reticulum. Biochimie. 2005;87:111–116. doi: 10.1016/j.biochi.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 41.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]