

Abstract
The cardiac neural crest migrate from rostral dorsal neural folds and populate the branchial arches, which directly contribute to cardiac-outflow structures. Although neural crest cell specification is associated with a number of morphogenic factors, little is understood about the mechanisms by which transcription factors actually implement the transcriptional programs that dictate cell migration and later the differentiation into the proper cell types within the heart. It is clear from genetic evidence that members of the paired box family and basic helix-loop-helix (bHLH) transcription factors from the twist family of proteins are expressed in and play an important function in cardiac neural crest specification and differentiation. Interestingly, both paired box and bHLH factors can function as dimers and in the case of twist family bHLH factors partner choice can clearly dictate a change in transcriptional program. The focus of this review is to consider the role that the protein-protein interactions of these transcription factors may play determining cardiac neural crest specification and differentiation and how genetic alteration of transcription factor stoichiometry within the cell may reflect more than a simple null event.
Cardiovascular development and the role of the cardiac NC
Neural crest cells become specified within the dorsal lip of the neural tube after which they migrate ventrally along the anterior posterior axis of the developing embryo and contribute to the formation of a variety of different tissues and organs including the skin, bone, neurons, and of course structures within the cardiovascular system. Deficiencies in cardiac neural crest cell (NC) development result in defective remodeling of the aortic arch arteries and failure of outflow tract septation. Development of a normally functioning heart and correctly remodeled vascular arterial system is required for embryonic viability (Winnier et al., 1999; Koushik et al., 2001; Conway et al., 2003), and the abnormal development of either of these aspects of the cardiovascular system can result in congenital heart defects that may be lethal or have costly lifelong effects.
The importance of neural crest cell populations to the proper development of the heart and associated vasculature is evident from classical cell ablation studies done in the chick. In these experiments, ablation of neural crest migrating from rhombmeres 6-8 result in a number of congenital heart defects which include persistent trunks arteriosus (PTA), double outlet right ventricle (DORV), tetralogy of fallot (TOF), and ventricular septal defects (VSD) (Kirby, 1999). These experiments are difficult to perform in a mammalian system; however, the power of genetics in the mouse has provided for experimental approaches that have complimented the chick data. The identification of existing mutant mouse models, the creation of gene knockouts via homologous recombination, and the employment of conditional marker gene activation using Cre-recombinase based expression systems have allowed both the fate mapping and study of gene-ablation of transcription factors expressed within the neural crest populations that contribute to the forming heart (Espstein, 2001). Although these studies have added great insight into the importance of neural crest in the formation of the heart, little is understood about the mechanisms by which transcription factors orchestrate the execution of the transcriptional programs of specification and differentiation, which are initiated by the extracellular signals instructing the cells.
Remodeling of the transitory symmetrical embryonic aortic arch arteries into the definitive adult left-sided aortic arch vascular pattern involves the asymmetrically programmed regression and persistence of specific arch arteries (see Fig. 1). The final arrangement and morphology of these great vessels requires reciprocal signaling between the endothelial cells lining the pharyngeal arch arteries (Yanagisawa et al., 1998), the surrounding NC-derived smooth muscle and mesenchyme (Le Lievre & Le Douarin, 1975; Fishman & Kirby, 1998) and the endoderm (Wendling et al., 2000; Vitelli et al., 2002). Additionally, the recently identified anterior heart-forming field gives rise to myocardium within both the outflow tract (Kelly & Buckingham, 2002) and right ventricle (Cai et al., 2003). It is unclear whether anomalies within this extra-cardiac cell population can also influence NC morphogenesis. Regardless on the effects on NC, this cell population is likely to play a role in the pathogenesis of outflow tract defects. Anomalous remodeling underlies a wide variety of congenital heart defects including: PTA; coarctation and interruption of the aortic arch; double aortic arch; right aortic arch and abnormal origin of the right subclavian. PTA arises when the arterial trunk fails to be divided to form a separate pulmonary artery and aorta. DiGeorge patients (1/4000 live births) - who can exhibit hypocalcemia, defective thymic-dependent cellular immunity, abnormalities of the face, ears and palate, interrupted aortic arch, outflow tract defect or some degree of overriding aorta - often have abnormalities of the NC-derived 3rd, 4th and 6th aortic arches (Epstein & Buck, 2000). Most patients are hemizygous for a deletion of chr.22q11. The genes within the ‘minimal critical region’ are known and under intense study, moreover several play a role in outflow tract development (Momma et al., 1999; Lindsay & Baldini, 2001). In contrast, similar deletions in mice have so far failed to completely recapitulate the syndrome, suggesting DiGeorge is multifactorial and that the contribution of the NC to the different aberrant tissues is complex, and that there may be other downstream genes that are important effectors (Epstein & Buck, 2000). Despite the presence of many different mouse mutant models of anomalous aortic arch artery development (several within the DiGeorge region), the precise role of the cardiac NC within cardiovascular development is not well understood (Olson & Srivastava, 1996; Maschhoff & Baldwin, 2000).
Figure 1. Analysis of cardiac neural crest cell migration and cardiac neural crest cell-deficient cardiac phenotypes.
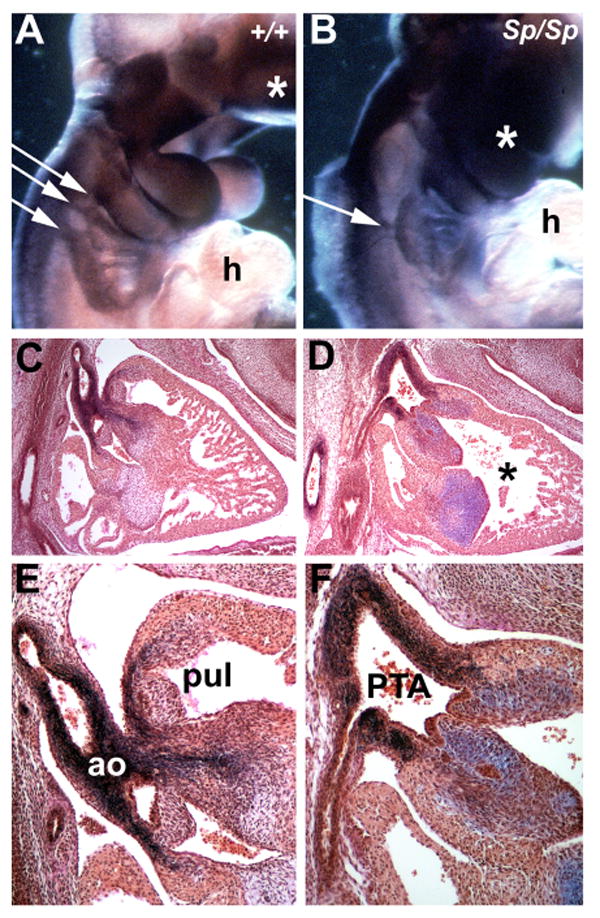
(A and B) Non-radioactive wholemount in situ analysis of the expression of the neural crest cell marker (cellular retinoic acid binding protein-1, CRABP1) in both wildtype (+/+) and pax3 muatnt (Sp2H/Sp2H) E10.5 embryos. Note that CRABP-1 is normally expressed within the craniofacial region of E10.5dpc Sp2H/Sp2H mutant embryos (indicated by *) but is mis-expressed within the cardiac neural crest cell region in Sp2H/Sp2H mutant embryo. Instead of the normal three streams of migrating neural crest cells (indicated by 3 small white arrows in +/+), there is only a single stream of migrating neural crest cells in the mutant embryo (indicated by single small white arrow in mutant). (C to F) Resorcin-fuchsin with van Gieson counterstain of E13.5 wildtype and Sp2H/Sp2H hearts. Note that in the low power image of normal heart (C), that there are two outflow tract vessels exiting the heart - the aorta (ao) and pulmonary trunk (pul) – and that each vessel exits a separate ventricular chamber divided by the interventricular septum. However, only a single vessel exits the Sp2H/Sp2H) heart, and that the interventriculr septum is incomplete (indicated by *). Additionally, notice that the elastin fibers (stained black) are reduced in number and do not enter the Sp2H/Sp2H heart and that there is excessive staining of the mucin glycoproteins (stained purple) in these mutant endocardial valve structures (F)
Cardiac NC is formed within the posterior rhombencephalon neural folds at the junction of the neuroectoderm and ectoderm (LaBonne & Bronner-Fraser, 1999) and between otic placode and 4th somite (reviewed by Creazzo et al., 1998). In the 8.5dpc mouse, they undergo epithelial-mesenchymal transformation and migrate ventrally along specific pathways into the 3rd, 4th and 6th pharyngeal arches. Arches are originally composed of mesenchyme (mesodermally-derived), which is surrounded externally by ectoderm and internally by endoderm. Subsequent to cardiac NC colonization, most of the mesenchyme is NC-derived, apart from the original mesodermal core that lies adjacent to the aortic arch arteries (Jiang et al., 2000). NC-derived mesenchyme subsequently condenses and differentiates into a fibrous connective tissue that contributes to vascular stabilization of the great arteries (Maschhoff & Baldwin, 2000), while other NC populate the cardiac ganglia (Waldo et al., 1999a). There is extensive experimental evidence (little of which is genetic) suggesting the various steps in NC morphogenesis are influenced by cell-cell and cell-matrix adhesions (reviewed by Erickson & Perris, 1993; Henderson & Copp, 1997). However, the role and influence of the NC once they arrive in the arches remains unclear. Cell fate studies using a transgenic cre/loxP cell marking technique have demonstrated that the mere presence or absence of NC is not sufficient to cause remodeling, as there does not appear to be any differences in the distribution between those arteries that persist verses those that regress (Jiang et al., 2000). This suggests that different left-right signals required for remodeling must be carried by the colonizing NC or are present within the mesodermal core, endothelial cells lining the arch arteries or within the pharyngeal pouch/cleft endoderm. The origin and identity of these signal/s is currently unknown, but may involve retinoic acid levels/signaling (Iulianella & Lohnes 2002) or differential activity of ion channels (polycystin-2; Pennekamp et al., 2002) giving rise to unidirectional transfer through gap junctions, resulting in asymmetric gene expression (reviewed by Mercola & Levin, 2001). Currently, pitx2 (Liu et al., 2001) is the only known gene asymmetrically expressed within the pharyngeal arch mesoderm, and it has been reported that an isoform-specific deletion of pitx2c results in abnormal patterning of the aortic arch vessels (Liu et al., 2002). Some cardiac NC continue to migrate on into the outflow tract, where they populate the conotruncal cushions (Creazzo et al., 1998; Conway et al., 1997a,b). Once in the outflow tract cushions, it has been suggested the cardiac NC are involved in fusion and subsequent myocardialization of the proximal outflow tract, becoming the muscular outlet septum of the heart (van den Hoff et al., 1999). NC from this same region of the mouse neural tube also gives rise to cells of the thymus, thyroid and parathyroid glands (Jiang et al., 2000; Epstein et al., 2000). Although the role of the cardiac NC remains mostly descriptive, some insights into the cellular processes have come from experimental manipulation of chick embryos (reviewed in Creazzo et al., 1998) and the genetic processes from mouse, zebrafish and Xenopus mutants (reviewed in Srivastava, 2001). In chick embryos, neural fold/NC ablation (Creazzo et al., 1998), Hox antisense experiments (Kirby et al., 1997), teratogenic retinoic acid exposure (Broekhuizen et al., 1998) and haemodynamic perturbations (Hogers et al., 1999) have all caused 4th and 6th arch abnormalities. Similarly in mouse, teratogenic exposure to haloacetic acids (Hunter et al., 1996), ethanol (Sulik et al., 1986), and retinoic acid (RA) gives rise to arch abnormalities. Related or analogous pathological defects of the mouse 4th and 6th aortic arches and outflow tract are also seen when a large number of genes have been transgenically altered.
Coordinated NC-derived mesenchyme differentiation is required for remodeling of the arterial tree and outflow tract septation
Mutational analysis has identified a large number of genes required for morphogenetic and inductive processes involving the mouse cardiac NC (reviewed in Creazzo et al., 1998; Epstein & Buck, 2000; Srivastava, 2001). These genes appear to involve several different pathways acting in parallel or in series with one another, but there is currently no simple pathway that unifies all the available data. Some genes, including pax3, are expressed in the migrating cardiac NC as they relocate to their positions around the great vessels and outflow tracts and differentiate into smooth muscle and connective tissue (Conway et al., 1997a & Conway et al., 2000). More recent evidence suggests that these interactions, and the programming of the cardiac NC is mediated by transcription factors including Mf1/Mfh1, HAND1/HAND2 and Tbx1/other genes on human chromosome 22q11 (Winnier et al., 1999; Srivastava, 2001; Merscher et al., 2001). Another set of genes is expressed within the developing vasculature itself, and may play a role in vessel formation, stabilization and remodeling (VEGF-A, Carmeliet et al., 2000; neuropilin-1, etc.) or within the interacting mesenchymal cells (MEF2C, Lin et al., 1998; tissue factor, Carmeliet et al., 1996 etc.) leading to the idea that the control over vascular assembly resides within the connective-tissue forming NC-derived mesenchyme. In support of these reciprocal interactions, Noden (1989) found similar results using quail-chick transplantation. Significantly, VEGF120/120 & 188/188 mutants (Stalmans et al., 2003) have a variety of complex aortic arch defects and about 50% of the VEGF188/188 embryos die in utero due to a complete lack of vasculature prior to NC migration, but ∼50% of the VEGF120/120 mutants either lack a ductus arteriosus (failure of left 6th to persist) and the descending aorta is abnormally positioned on the right or they have a double arch (failure of right 4th to be remodeled). Both in vitro and in vivo analysis of NC indicates that NC formation and migration are not responsible for generating the mutant phenotypes, but that expression of VEGF120/164188 within the aortic arch endoderm is required for attraction of neuropilin-1 positive cells (both endothelial & NC-derrivied) to both stimulate ingrowth of arch arteries and for their maintenance (Stalmans et al., 2003). Finally, there is still another set of genes which is expressed in the pharyngeal arches themselves, and may play a role in mediating interactions between the arch epithelia (ectoderm and endoderm), mesenchyme and endothelial vessels (endothelin-1; Kurihara et al., 1995; Ece-1, Clouthier et al., 1998; EtA, Yanagisawa et al., 1998; semaphorin3c, Feiner et al., 2001). Thus, it is well established that cardiac NC can provide structural integrity and may also carry instructive signals required for aortic arch remodeling. Disruption of this signaling leads to defects in the interactions between post-migratory cardiac NC and the endothelium of the great vessels and outflow tract. Collectively, these results support a model in which epithelia/endothelia of the arches signals to NC-derived mesenchyme (possibly via processed ET-1, semaphorin and/or VEGF) maybe through gap junctions. However, it is unclear exactly what the function of the NC is within the arches and outflow tract, how they differentiate into connective tissue, how the wide array of transcription factors function in unison and which genes respond to the various NC-mediated differentiation signals.
Understanding the Role of Transcriptional Regulation within the Cardiac Neural Crest
Although there are a number of transcription factors known to be expressed in cardiac neural crest cells that populate the heart and mutations/ablation of expression of these genes result in phenotypic defects, the molecular mechanisms that drive these phenotypic outcomes is largely unknown. It is also evident that many of the transcription factors that are shown to be expressed and in some cases play a role in proper neural crest specification, expansion, migration and differentiation are also expressed in non-neural crest derived cells. To date, only eye and skeletal muscle cell specification and differentiation is associated with the expression of “master regulatory” transcription factors that when expressed in ectopic environments can in isolation give rise to fully formed eyes (Halder et al., 1995; Wawersik & Maas 2000) or in a tissue culture can initiate the skeletal muscle program (Olson, 1993; Olson and Klein, 1994). The cell specification and differentiation of remaining embryonic tissues most likely rely on combinatorial interactions between multiple tissue-restricted and general transcription factors that collectively through precise interactions form unique transcriptional complexes that define and drive the genetic programs of cell types such as cardiac neural crest (Firulli and Olson, 1997). In many cases such as transcription factors in the basic helix-loop-helix (bHLH) family of proteins, the nature of their interactions (dimerization) is obvious. In other examples such as the Pax transcription factors, the dependence on protein-protein interactions is not as nearly as clear; however, as Pax proteins contain both a paired domain and a helix-turn-helix motif, DNA binding to different cis-target sequences is likely a key regulatory switch for activating specific transcriptional programs. If the combinatorial model of tissue specific gene expression holds true, then a central question one must consider is: Are the resultant phenotypes observed from a “null” allele truly a reflection of the loss of that one protein? In order to gain a greater understanding of the molecular mechanisms that drive the cell specification and the differentiation of the cardiac neural crest, a step back must be taken to consider the transcription factors expressed with the cardiac neural crest as well as the biochemical properties that are essential for there function. Questions such as which transcription factors are coexpressed within the neural crest interact? What are the post-translational modifications, which alter transcriptional activity/localization/dimerization bringing the factors in and out of play? And finally, how does alteration of the precise transcription factor stoichiometry alter the multi-protein transcriptional complexes driving tissue identity. What follows is a review of what is understood about the mechanistic function of Pax and the HAND subclass of bHLH transcription factors that is focused on the function of characteristics of these proteins and the speculation on how alteration of these functional properties can result in congenital heart disease.
The Paired-box transcription factors
One of the most well studied transcription factor families implicated for the proper specification and differentiation of the cardiac NC is the Paired box (Pax) transcription factors. The nine Pax proteins are a family of transcriptional regulators involved in many developmental processes in all higher eukaryotes. The Pax3 and Pax7 sub-family are unusual as they encode proteins with two DNA-binding domains, a paired domain and a homeodomain. The paired domain is a bipartite DNA binding domain that is composed of two helix-turn-helix motifs, while the homeodomain itself is able to form both homo- and hetero-dimers on DNA. Thus, pax3 and pax7 proteins use multiple combinations of the three separate motifs to recognize DNA and effect transcriptional control (Jun & Desplan, 1996; Miskiewicz et al., 1996). The homeodomain is important in binding of DNA and in effecting transcriptional control. Thus, an absence of Pax proteins is likely to lead to wide-ranging abnormalities and mutations within any of the DNA-binding motifs is likely result in structural change within the homeodomain that either changes the DNA-binding specificity of the homeodomain or reduces the affinity of the pax3 protein for DNA. Furthermore, Pax7 and Pax3 are differentially methylated; as the gene region that encodes the paired domain is hypomethylated, whereas the region that encodes the homeodomain is hypermethylated (Ziman & Kay, 1998).
The phenotypic consequences of misregulated pax3 are well understood however the actual molecular pathways, which are disrupted in pax3 mutations, are not well defined. There are a number of pax3 mutations within the mouse that are collectively termed splotch mutants. There are 5 splotch alleles with different mutations of the pax3 transcription factor: Two of these alleles (sp and sp2H) have provided a robust and morphologically well-characterized model of aortic arch and outflow tract defects (reviewed in Epstein & Buck, 2000; Conway et al., 1997a & b; Conway et al., 2000; see Fig 1). However, the molecular mechanisms underlying the pathogenesis of the defects remains largely unknown - primarily due to the lack of known downstream effector genes and an understanding of what role pax3 actually plays within the NC cells themselves. Pax3 is unusual among the Pax family members because it is thought to be predominantly a negative regulator of target genes leading to suppression of target gene expression (Chalepakis et al., 1994a). This suggests downstream targets of pax3 are likely to be repressed under normal circumstances, and sp2H mutant Pax3 protein will result in aberrant over-expression. The different splotch alleles disrupt the pax3 gene in varying ways (reviewed by Chalepakis et al., 1994a,b), and it seems likely that each mutation results in loss of some or all of the functions of the pax3 gene in controlling downstream gene expression.
The sp2H homozygotes (Franz 1989; Conway et al., 1997a,b,c) die in utero due to the presence of PTA with obligatory perimembranous VSD. The pathogenesis of the defect is due to the failure of the left 6th arch artery to persist, which usually gives rise to the pulmonary trunk. Thus, the abnormal regression of the left 6th aortic artery is the first anatomical defect present within the sp2H mutant cardiovascular system, and is the site at which the consequences of abnormal cardiac NC development is first evident. In addition, sp2H embryos exhibit defects within neural tube closure (spina bifida & excencephaly), melanocytes and lack of limb musculature (Dickman et al., 1999; Schubert et al., 2001). Significantly, pax3 mRNA is expressed within the neural tube, migratory NC cells and their derivatives (such as the thymus, thyroid & dorsal root ganglia), somites and melanocytes (Goulding et al., 1993) – all structures abnormal in sp2H homozygotes. In humans, PAX3 mutations lead to Waardenburg syndrome, an autosomal dominant disorder that consists of defects in NC-derived tissues and is characterized by pigmentation, hearing and facio-skeletal anomalies (Tassebehji et al., 1995). Cardiac defects have also been reported in Waardenburg children (Banerjee, 1986; Mathieu et al., 1990). The sp2H mutants contain a 32bp deletion in the homeodomain, one of the two main DNA-binding regions of the pax3 gene (Epstein et al., 1991). This deletion leads to formation of a stop codon such that the C-terminal portion of the homeodomain is missing from the mature protein. DNA-binding studies in vitro show that a protein with a structure similar to the predicted truncated sp2H Pax3 protein has reduced homeobox DNA-binding activity with altered specificity for target sequences (Chalepakis et al., 1994a). As the paired box is unaltered, sp2H Pax3 preferentially bind the paired box consensus (GTTCC). Moreover, as dimerization of pax3 can occur via the homeodomain the loss of the domain will likely alter important protein-protein interactions required for pax3-medieated gene expression.
Another splotch mutation is sp. Sp mutants have an A→T transversion in a splice acceptor site, which results in four aberrant transcripts (Epstein et al., 1993) that are not expected to produce functional Pax3 proteins and thus sp mutants are thought to be a null allele. This is supported by recent targeted deletion of pax3 that recapitulates the sp phenotype (Mansouri et al., 2001).
Although sp2H NC initiate migration, they do not reach the branchial arches or outflow tract in sufficient numbers for aortic arch remodeling or outflow tract septation to occur (Serbedzija & McMahon, 1997; Conway et al., 1997a; Epstein et al., 2000). Furthermore, it has been shown that this lack of colonizing cardiac NC is due to abnormal NC stem cell expansion (Conway et al., 2000). Additionally, there is low-level pax3 expression in normal 9.5dpc hearts (Conway et al., 1997a) and that 100% of the homozygous mutant embryos die by 14.0dpc solely due to the presence of PTA/VSD anomalies (Conway et al., 1997a,b) and not the neural tube defects. Surprisingly, deficiencies in myocardial function further compromise cardiac function, as there is abnormal excitation-contraction (EC) coupling in the sp2H mutants (Conway et al., 1997b) and an abnormally thinned myocardium in the sp mutants (Li et al., 1999), resulting in in utero lethality by ∼14dpc. It has been proposed that this myocardial defect is an indirect consequence of the reduced numbers of migrating NC (Kwang et al., 2002), because NC cells are not thought to contribute to the myocardium (Jiang et al., 2000). However, Kirby and colleagues have shown that pre-migratory cardiac NC ablation in chick results in myocardial dysfunction prior to the arrival of the cardiac NC within the heart, and have suggested that these early effects on the heart are due to a prolonged release of factors (FGFs etc.) by the pharyngeal endoderm, which are normally involved in the induction of cardiac mesoderm (Waldo et al., 1999b). Additionally, expression of the Lbx1 homeobox gene that is expressed in a subpopulation of the cardiac NC is upregulated in the hearts of Lbx1 null/sp1H/sp1H (sp1H is allelic with sp2H) mice, indicating that Pax3 and Lbx1 participate in a negative regulatory feedback that might be necessary for normal differentiation and function of the myocardium during early heart development (Schafer et al., 2003). Thus, it is interesting to note that pax3 descendent cells (marked via transgenic Cre/loxP and ROSA26R techniques) give rise to populations of cells present within the E13 mouse heart (Li et al., 2000). Finally, cre/loxP deletion of the BMP receptor 1A in cardiac NC reveals that bmp2/4 signaling in NC derivatives is essential for outflow tract development and may regulate a crucial proliferation signal for the ventricular myocardium (Stottmann et al., 2004). Thus, although it is clear that cardiac NC deficiency leads to defective remodeling of the aortic arch arteries and failure of outflow tract septation, it will be interesting to determine the precise role that cardiac NC cells play within the heart itself.
Significantly, pax3-FKHR knockin heterozygous mice are not viable and all die around birth due to cardiac insufficiency (Lagutina et al; 2002). Alveolar rhabdomyosarcoma is associated with chromosomal translocation t(2;13) causing a fusion of PAX3 (& PAX7) on chromosome 2 to the FKHR gene on chromosome 13 (Anderson et al., 2001). The resulting fusion proteins contain the N-terminal region of pax protein fused to the potent C-terminal transactivation domain of FKHR. Studies suggest that the tumor-associated pax3-FKHR protein can interfere with normal pax3 developmental functions (Anderson et al., 2001), and analysis of the pax3-FKHR knockin heterozygotes reveales that they have a VSD, a grossly enlarged septum and a dilated outflow tract and die at birth, but do not develop tumors (Lagutina et al., 2002). Also the fetal liver was engorged with blood and the lungs failed to inflate, all classic signs of cardiac failure (Conway et al., 1997c). Thus, it is currently unclear if the pax3-FKHR knockin is acting as a ‘dominant-negative’ mutation affecting the heart (as pax3 itself has not been thought to play a role in myocardial morphogenesis) or if the mutation in pax3 directly affects the heart (as pax3 mRNA was detectable within 10.5dpc pharyngeal arches and Pax3 protein was detectable within the 16.5dpc heart). Although not identical, it is interesting to note that both the pax3-FKHR and sp2H mutants lack the C-terminal portion of the homeodomain that modulates homeobox DNA-binding activity/dimerization and thus may modulate pax3 specificity for its distinct target sequences (Chalepakis et al., 1994b). Given these combined data, it will be interesting to determine whether there is a primary and/or secondary role for the cardiac NC during cardiomyocyte morphogenesis using conditional gene targeting and to deduce the functional significance of the loss of the homeodomain on NC-specific transcription.
The sp cardiac phenotype can be rescued by transgenic over-expression of pax3 under the control of a 1.6kb ‘neural tube/NC-specific’ Pax3 promoter that is not expressed within the somites (Li et al., 1999). Transgenic/sp homozygotes survive until birth, at which time they succumb to respiratory failure secondary to absence of a muscular diaphragm, and limb muscles were also absent. Pax3-deficient somites are capable of supporting proper NC migration and function indicating a cell autonomous role for pax3 in NC morphogenesis. However, Gruss and colleagues (Mansouri et al., 2001) used ES cells to generate a lacZ knockin into the pax3 locus. The pax3 knockin allele was used to generate pax3-deficient ES cells to investigate whether, in chimeric embryos (using sp2H rather than sp), pax3 acts cell autonomously. Pax3 function was shown to be essential for the neuroepithelium and somites, but a wild-type environment rescued pax3 knockin null NC cells, suggesting pax3 does not act cell autonomously during NC migration.
Although several laboratories have proposed likely downstream targets for regulation by pax-3, only one has proved to play an in vivo role in the pathogenesis of heart defects in splotch mutants. Surprisingly, loss of msx2 rescues the sp cardiac defects (i.e. ‘two wrongs (nulls) make a right’), as well as defects in the dorsal root ganglia, thymus and thyroid (Kwang et al., 2002). Both the endogenous msx2 gene and transgenes comprising fragments of the msx2 promoter-lacZ reporter are upregulated in sp homozygous mutants, suggesting that msx2 is negatively regulated by pax3. Mutation of a pax3-binding site within the msx2 promoter increased msx2 transgene expression and provided evidence that pax3 is a direct transcriptional repressor of msx2. Interestingly, the expression domains of pax3, msx1 and msx2 overlapped only within the neural tube and not within the aortic arches or heart itself, as only msx2 is expressed within the developing heart – particularly the conduction system (Chan-Thomas et al., 1993). Msx1 nulls indicate that msx1 is required for development of the molar tooth and palate (Chen et al., 1996) and lack of msx2 results in defects in calvarial bones, tooth, skin and mammary gland development – but neither mutant has any reported cardiovascular defects. Msx1/2 double homozygous mutants die in utero ∼16dpc, with severe defects in cranial NC development but no reported heart defects (Satokata et al., 2000). Thus, the mechanism by which loss of msx2 rescues the sp cardiac defects is unknown, but presumably pax3/msx2 are jointly required to control NC expansion/differentiation of the NC population within the neural tube, as over-expression of msx2 has been shown to result in hyperproliferation (Wang et al., 1999) and inhibition of differentiation (Dodig et al., 1999). An alternate hypothesis would be that pax3 and msx2 could be co-regulators of common transcriptional targets where mutations in pax3 could result in deleterious regulation of these genes due to inappropriate interactions. Loss of msx2 simply removes the ability to encounter these deleterious interactions. Assuming that pax3 and msx2 are not necessary for target gene activation, this model would fit well and if true suggests that mutant pax3 interactions with other wildtype proteins mediate many of the observed phenotypic outcomes.
HAND bHLH transcription factors
HAND factors fall within the B class of the bHLH proteins and are most highly related to twist-like bHLH genes (for review (Firulli, 2003)). Like all bHLH proteins HAND factors function as dimers to bind a canonical sequence CANNTG termed an E-box (Dai and Cserjesi, 2002; Hollenberg et al., 1995). HAND1 and HAND factors are expressed within a number of tissues during mouse development which include the lateral mesoderm, extraembryonic mesoderm, maternally derived decidua, heart and cardiac neural crest that populate the branchial arches and outflow tract (Firulli, 2003). Both HAND1 and HAND2 have been knocked out in the mouse (Firulli et al., 1998; Riley et al., 1998; Srivastava et al., 1997). The effect of the loss of HAND1 on neural crest cannot be deduced as HAND1 mice die between E8.0 and E9.0 (Firulli et al., 1998; Riley et al., 1998). HAND2 null mice die between E9.5 and E10.5 and exhibit, hypoplastic first and second arches secondary to apoptosis and the third and fourth arches fail to form (Srivastava et al., 1997; Thomas et al., 1998).
Molecular analysis of HAND2 -/- mice reveal that the majority neural-crest-derived components of the branchial arch are expressed, suggesting that normal migration of neural crest cells occurs. Expression of HAND1 and HAND2 within the neural crest cells populating the branchial arches is in part regulated by Endothelin-1 (ET1) signaling (Clouthier et al., 2000; Thomas et al., 1998). ET-1 -/- mice phenotypically are similar to traits observed in humans with CATCH-22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia, associated with chromosome 22 microdeletion) and show a downregulation of both HAND1 and HAND2 within the branchial arches (Clouthier et al., 2000; Thomas et al., 1998). ET1 regulation of HAND factors is evolutionarily conserved and from HAND2 transcriptional regulation studies in the mouse, HAND2 transcription is mediated by the actions of the Distal-less class homeodomain transcription factor Dlx6 (Charite et al., 2001; Miller et al., 2000). Targeted disruption of the HAND2 brachial arch enhancer results in loss of HAND2 expression within the first and second brachial arch resulting in a number of craniofacial abnormalities (Yanagisawa et al., 2003). It should be noted that HAND2 expression in the ventral domain of the branchial arch is maintained in this mouse model as well as HAND2 expression within the cardiac neural crest that populate the outflow tract (Yanagisawa et al., 2003). Further dissection of HAND2 promoter or generation of HAND1 and HAND2 conditional alleles will be required for examination of HAND “null” phenotypes of the cardiac NC; however based on the defect observed due to deletion in the craniofacial NC, defects in cardiac neural crest are likely to be encountered.
In addition to the developmental studies, insights into the molecular mechanisms have been reported. Expression of HAND1 and HAND2 within avian neural crest cells can dive these cells into catechlolaminergic neurons (Howard et al., 1999). More recently HAND factor dimerization control has been associated with alteration in HAND transcriptional program and this dimerization control is likely to play a role in the proper specification and differentiation of the cardiac NC (Firulli et al., 2003). The original model for bHLH function deduced from studies of the myogenic bHLH proteins was that a tissue-restricted or class B bHLH factor partnered with a ubiquitously expressed class A or E-protein to bind DNA and modulate transcription (for review see (Massari and Murre, 2000)). Although this model certainly holds true, HAND1 and HAND2 exhibit a promiscuous ability to dimerize with themselves and other class B bHLH factors (Firulli et al., 2003; Firulli et al., 2000). When one considers HAND expression and the co expression of other bHLH proteins, an expanded pool of possible HAND dimers becomes evident. This further suggests that the bHLH dimer pool within the cardiac NC may be carefully balanced such that some HAND dimers may function as activators in one genetic program while other HAND dimers could simultaneously function as repressors. This hypothesis begs the question what are the molecular mechanisms (other than expression) that facilitate HAND dimerization choices within the cardiac NC? Posttranslational modifications such as phosphorylation, can dramatically change function of bHLH DNA binding (Li et al., 1992a; Li et al., 1992b) and homocycteine disulfide linkages have been show stabilize E-protein homodimers in blood cell development (Benezra, 1994; Markus and Benezra, 1999). In our recent efforts, we show that HAND factor dimerization is in part regulated by the phosphorylation of a threonine and serine located in helix I of the bHLH domain (Firulli et al., 2003). These residues are phosphorylated by PKA and PKC. Dephosphorylation of these residues is mediated by the protein phosphatase 2A (PP2A) that specifically contains the B56δ regulatory subunit. Experimental data show that coexpression of HAND1 with constitutively active PKA increases specific phosphorylation on a number of peptides. Where coexpression of the non-HAND interacting PP2A regulatory subunit B56α, shows no effect on HAND1 phosphorylation, coexpression of B56δ with PKA and HAND1 results in the specific dephosphorylation of HAND1 (Firulli et al., 2003). Similar findings are observed in the phosphorylation of HAND2. The functional consequences of phosphorylation on these residues are altered dimerization affinities with prospective bHLH partners (Firulli et al., 2003). Experiments performed in the developing chick limb show that HAND1 biological function is effected by alteration of these key regulator residues as both hypo- and phosphorylation mutants in HAND1 drive distinct phenotypic outcomes from wild type HAND1 expression within the limb (Firulli et al., 2003).
Although the HAND phosphorylation state relating to cardiac NC development has not been studied, it is likely that HAND factors will be regulated in a similar manner during cardiac NC specification and differentiation. To address the issue of which HAND- dimer pairs are important for cardiac NC specification and differentiation the use of a variety of tethered HAND-dimer constructs could be employed in expression screens and knockin rescue experiments in hopes of adding insight to the identity of direct HAND downstream genes. Such future studies are likely underway and will allow for a more detailed understanding of the role HAND factors play in the cardiac neural crest cell population.
If transcription factors interact, what is a null phenotype?
An important consideration when trying to understand the phenotypic outcome of mouse models with NC-derived cardiac defects is the root cause of the phenotype. In the case of a transcription factor, the obvious cause is the disruption of transcriptional control of downstream target genes. If the transcription factor in question is not expressed, it will not bind DNA and will not activate/repress the genes under its regulation. When considering bHLH proteins, the scenario is more complex (Fig 2B). Considering the promiscuous dimerization profiles exhibited by HAND1 and HAND2 gene deletion of a HAND factor not only eliminates its direct participation in the transcriptional regulation of its downstream targets, its absence also results in a shift in bHLH factor stoichiometry within the cell that can result in the formation of inappropriate dimers between the remaining bHLH proteins within the pool. These dimers could inappropriately regulate HAND down-stream targets or could activate alternative genetic programs causing phenotypic mishaps more extensive than a simple null phenotype. For example, consider the HAND2 branchial arch enhancer knockout where HAND2 expression is reduced in both the first and second branchial arch (Yanagisawa et al., 2003). In addition to E-proteins the bHLH factor HAND1 and m-twist are partially coexpressed with HAND2 within the first branchial arch. The loss of HAND2 in the first arch changes the equilibrium between HAND1, Twist1 and E-factors driving the formation of a new dimer pool by mass action were these HAND1,Twist1 and E-protein homo and heterodimers will modulate different transcriptional instructions that can result in cell death, respecification, and or decreased/increased growth. This model is supported by reports that Twist1 can function as a homodimer and E-protein heterodimer and that Twist1 and HAND1 and HAND2 can form heterodimers when coexpressed (Castanon et al., 2001); (ABF unpublished).
Figure 2. Spaciotemporal expression of HAND1 and HAND2 in the branchial arch mesenchyme and heart.
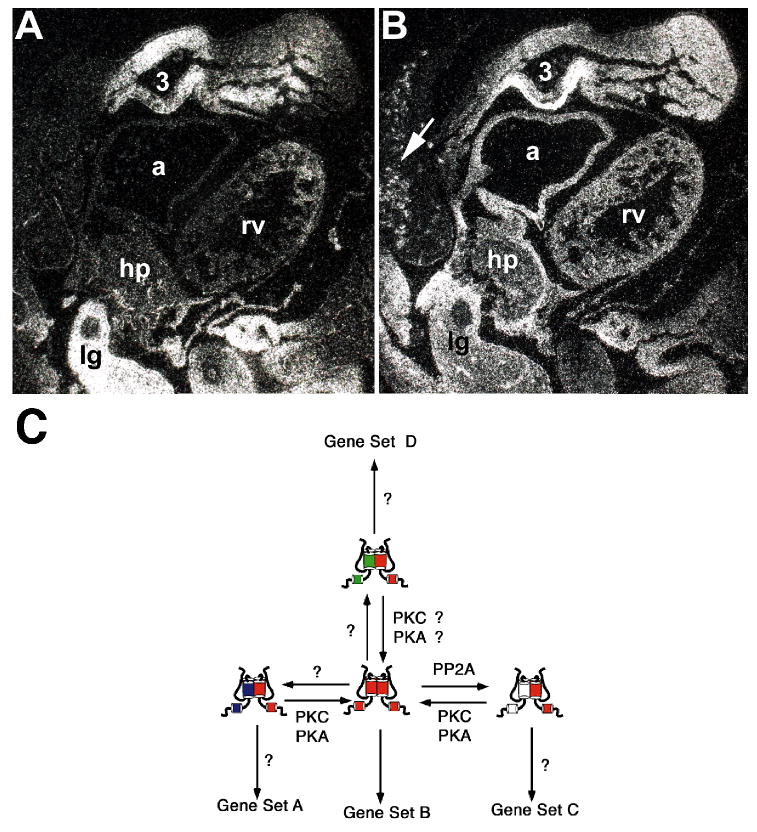
(A) HAND1 mRNA expression within the E10.5 mouse embryo as detected by radioactive in situ hybridization. Note that HAND1 is expressed within the mandibular mesenchymal arch core of the first branchial arch, the third arch, the nascent interventricular septum of the ventricles but not within the right ventriucle (rv) and atria (a). Additionally, HAND1 is highly expressed within the hepaticprimordia/septum transversum (hp) and lung bud (lg). (B) HAND2 mRNA expression within the E10.5 mouse embryo as detected by radioactive in situ hybridization. Note that HAND2 is co-expressed with HAND1 within the mandibular region of the first branchial arch, the third arch, the hepaticprimordia/septum transversum and lung bud; but, unlike HAND1 it is expressed throughout the right ventricle and atria. Additionally, punctate putative HAND2-positive neural crest cells are present within the brachial arch and trunk mesenchyme (arrow), which are not seen with the HAND1 probes. (C). Model of how HAND factor dimerization dictates tissue-specific gene expression. Regulation of the phosphorylation state of key residues in helix 1 by the actions of PKA and B56δ-containing PP2A and overall levels of gene expression drive HAND-Dimer formation. Different dimers juxtapose different basic domains, which bind different E-boxes and or act as activators or repressors thus driving tissue-specific transcriptional instructions.
When considering non-bHLH factors such as Pax3 similar mechanisms can also be applied. Furthermore considering that pax proteins can recognize 2 distinct cis-elements the paired and homeobox regulations of which cis-element is preferentially bound is also a key factor in define specific transcriptional response. Deletion of the Pax3 homeodomain removes pax3 from directly accessing homeodomain-dependent transcriptional targets. Protein-protein interactions are also disrupted allowing putative pax3 partners free of their interactions to regulate transcription in an inappropriate manner. If interactions with pax3 are not effected these factors may be titrated away from the promoters dependent on pax3 DNA binding. In the case of msx2 null mice crossed into the splotch background the cardiac defects observed are rescued. As msx2 (as well as msx1 msx2 double nulls) nulls show no overt cardiac phenotypes the rescue of splotch is difficult to understand without consideration of biochemical interactions. If expansion/differentiation of the NC population within the neural tube is not dependent on either msx or pax proteins but an abhorant interaction between mutant pax3 and msx can inhibit these If NC expansion/differentiation, one could explain how loss of msx2 could rescue these developmental defects.
In conclusion it is clear that much is understood on the role that pax3 play in the proper specification and differentiation of the cardiac NC however the molecular mechanism that drives cardiac NC disregulation is not well understood. Conversely the role of HAND1 and HAND2 in the cardiac NC is poorly understood yet there is a firm understanding of molecular regulation of how these factors drive tissue specific gene expression. To gain a more comprehensive understanding on how both pax3 and HAND factors fit into the cell specification and differentiation transcriptional program of cardiac NC both genetic and functional biochemical analysis of these factors must be combinatorial interactions between transcription factors must be concurrently pursued.
Acknowledgments
This work was supported by grants from the National Institutes of Health (2RO1HL61677-05) and March of Dimes Birth Defects Foundation (#1-FY02-276) (ABF). And supported in part by HL60714, HL33756 and HL52813 (S.J.C.)
References
- Anderson MJ, Shelton GD, Cavenee WK, Arden KC. Embryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc Natl Acad Sci U S A. 2001;98(4):1589–94. doi: 10.1073/pnas.98.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee AK. Waardenburg's syndrome associated with ostium secundum atrial septal defect. J R Soc Med. 1986;79:677–678. doi: 10.1177/014107688607901121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R. An intermolecular disulfide bond stabilizes E2A homodimers and is required for DNA binding at physiological temperatures. Cell. 1994;79:1057–1067. doi: 10.1016/0092-8674(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Broekhuizen ML, Gittenberger-de Groot AC, Baasten MJ, Wladimiroff JW, Poelmann RE. Disturbed vagal nerve distribution in embryonic chick hearts after treatment with all-trans retinoic acid. Anat Embryol (Berl) 1998;197(5):391–7. doi: 10.1007/s004290050150. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003 Dec;5(6):877–89. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Mackman N, Moons L, Luther T, Gressens P, Van Vlaenderen I, Demunck H, Kasper M, Breier G, Evrard P, Muller M, Risau W, Edgington T, Collen D. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383:73–5. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Castanon I, Von Stetina S, Kass J, Baylies MK. Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development. 2001;128:3145–3159. doi: 10.1242/dev.128.16.3145. [DOI] [PubMed] [Google Scholar]
- Chalepakis G, Jones FS, Edelman GM, Gruss P. Pax-3 contains domains for transcription activation and transcription inhibition. Proc Natl Acad Sci USA. 1994a;91(26):12745–9. doi: 10.1073/pnas.91.26.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G, Wijnholds J, Gruss P. Pax-3-DNA interaction: flexibility in the DNA binding and induction of DNA conformational changes by paired domains. Nucleic Acids Res. 1994b;22(15):3131–7. doi: 10.1093/nar/22.15.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Thomas PS, Thompson RP, Robert B, Yacoub MH, Barton PJ. Expression of homeobox genes Msx-1 (Hox-7) and Msx-2 (Hox-8) during cardiac development in the chick. Dev Dyn. 1993;197(3):203–16. doi: 10.1002/aja.1001970305. [DOI] [PubMed] [Google Scholar]
- Charite J, McFadden DG, Merlo G, Levi G, Clouthier DE, Yanagisawa M, Richardson JA, Olson EN. Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes & Development. 2001;15:3039–3049. doi: 10.1101/gad.931701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bei M, Woo I, Satokata I, Maas R. Msx1 controls inductive signaling in mammalian tooth morphogenesis. Development. 1996;122(10):3035–44. doi: 10.1242/dev.122.10.3035. [DOI] [PubMed] [Google Scholar]
- Chua CC, Hamdy RC, Chua BH. Mechanism of transforming growth factor-beta1-induced expression of vascular endothelial growth factor in murine osteoblastic MC3T3-E1 cells. Biochim Biophys Acta. 2000;1497(1):69–76. doi: 10.1016/s0167-4889(00)00040-9. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125(5):813–24. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Williams SC, Yanagisawa H, Wieduwit M, Richardson JA, Yanagisawa M. Signaling pathways crucial for craniofacial development revealed by endohelin-A receptor-deficient mice. Dev Biol. 2000;217:10–24. doi: 10.1006/dbio.1999.9527. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the (Sp2H) mutant. Development. 1997a;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Godt RE, Hatcher C, Leatherbury L, Zolotouchnikov VV, Brotto MAP, Copp AJ, Kirby ML, Creazzo TL. Neural crest is involved in development of abnormal myocardial function. J Molecular and Cellular Cardiology. 1997b;29:2675–2685. doi: 10.1006/jmcc.1997.0499. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Kirby ML, Anderson RH, Copp AJ. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovascular Res. 1997c;36:163–173. doi: 10.1016/s0008-6363(97)00172-7. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Abnormal neural crest stem cell expansion is responsible for the conotruncal heart defects within the Splotch (Sp2H) mouse mutant. Cardiovascular Research. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why. Genesis. 2003 Jan;35(1):1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of Cardiac Neural Crest Cells in Cardiovascular Development. Annual Review of Physiology. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- Dai YS, Cserjesi P. The basic helix-loop-helix factor HAND2 functions as a transcriptional activator by binding to E-boxes as a heterodimer. J Biol Chem. 2002;277:12604–12612. doi: 10.1074/jbc.M200283200. [DOI] [PubMed] [Google Scholar]
- Dickman ED, Rogers R, Conway SJ. Abnormal skeletogenesis occurs coincident with increased apoptosis in the Splotch (Sp2H) mutant--putative roles for Pax3 and PDGFRα in rib patterning. Anatomical Record. 1999;255:353–361. doi: 10.1002/(SICI)1097-0185(19990701)255:3<353::AID-AR11>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Dodig M, Tadic T, Kronenberg MS, Dacic S, Liu YH, Maxson R, Rowe DW, Lichtler AC. Ectopic Msx2 overexpression inhibits and Msx2 antisense stimulates calvarial osteoblast differentiation. Dev Biol. 1999;209(2):298–307. doi: 10.1006/dbio.1999.9258. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell. 1991;67(4):767–74. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, Vogan KJ, Trasler DG, Gros P. A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant. Proc Natl Acad Sci USA. 1993;90(2):532–6. doi: 10.1073/pnas.90.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JA, Buck CA. Transcriptional regulation of cardiac development: implications for congenital heart disease and DiGeorge syndrome. Pediatr Res. 2000;48(6):717–24. doi: 10.1203/00006450-200012000-00003. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127(9):1869–78. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Espstein JA. Devloping models of Digeorge syndrome. Trends Genet. 2001;17:13–17. doi: 10.1016/s0168-9525(01)02450-7. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Perris R. The role of cell-cell and cell-matrix interactions in the morphogenesis of the neural crest. Dev Biol. 1993;159(1):60–74. doi: 10.1006/dbio.1993.1221. [DOI] [PubMed] [Google Scholar]
- Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- Firulli AB. A HANDful of questions: The molecular biology of the HAND-subclass of basic helix-loop-helix transcription factors. Gene. 2003;312C:27–40. doi: 10.1016/s0378-1119(03)00669-3. [DOI] [PubMed] [Google Scholar]
- Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extraembryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nature Genetics. 1998;18:266–270. doi: 10.1038/ng0398-266. [DOI] [PubMed] [Google Scholar]
- Firulli AB, Olson EN. Modular regulation of muscle gene transcription: a mechanism for muscle cell diversity. Trends in Genetics. 1997;13:364–369. doi: 10.1016/s0168-9525(97)01171-2. [DOI] [PubMed] [Google Scholar]
- Firulli B, Howard MJ, McDaid JR, McIlreavey L, Dionne KM, Centonze V, Cserjesi P, Virshup DM, Firulli AB. PKA, PKC and the Protein Phosphatase 2A Influence HAND factor function: A Mechanisms for Tissue Specific Transcriptional Regulation. Mol Cell. 2003;12:1225–1237. doi: 10.1016/s1097-2765(03)00425-8. [DOI] [PubMed] [Google Scholar]
- Firulli BA, Hadzic DB, McDaid JR, Firulli AB. The basic helix-loop-helix transcription factors dHAND and eHAND exhibit dimerization characteristics that suggest complex regulation of function. Journal of Biological Chemistry. 2000;275:33567–33573. doi: 10.1074/jbc.M005888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman MC, Kirby ML. Fallen arches, or how the vertebrate got its head. J Clin Invest. 1998;102(1):1–3. doi: 10.1172/JCI4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz T. Persistent truncus arteriosus in the Splotch mutant mouse. Anat Embryol (Berl) 1989;180(5):457–64. doi: 10.1007/BF00305120. [DOI] [PubMed] [Google Scholar]
- Goulding M, Sterrer S, Fleming J, Balling R, Nadeau J, Moore KJ, Brown SD, Steel KP, Gruss P. Analysis of the Pax-3 gene in the mouse mutant splotch. Genomics. 1993;17(2):355–63. doi: 10.1006/geno.1993.1332. [DOI] [PubMed] [Google Scholar]
- Halder G, Callaerts P, Gehring WJ. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science. 1995 Mar 24;267(5205):1788–92. doi: 10.1126/science.7892602. [DOI] [PubMed] [Google Scholar]
- Henderson DJ, Copp AJ. Role of the extracellular matrix in neural crest cell migration. J Anat. 1997;191(Pt 4):507–15. doi: 10.1046/j.1469-7580.1997.19140507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogers B, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc Res. 1999;41(1):87–99. doi: 10.1016/s0008-6363(98)00218-1. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H. Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol Cell Biol. 1995;15:3813–3822. doi: 10.1128/mcb.15.7.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, Foster DN, Cserjesi P. Expression of HAND gene products may be sufficient for the differentiation of avian neural crest-derived cells into catecholaminergic neurons in culture. Developmental Biology. 1999;215:62–77. doi: 10.1006/dbio.1999.9450. [DOI] [PubMed] [Google Scholar]
- Hunter ES, 3rd, Rogers EH, Schmid JE, Richard A. Comparative effects of haloacetic acids in whole embryo culture. Teratology. 1996;54(2):57–64. doi: 10.1002/(SICI)1096-9926(199606)54:2<57::AID-TERA1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Iulianella A, Lohnes D. Chimeric analysis of Retinoic Acid Receptor Function during Cardiac Looping. Dev Biol. 2002;247(1):62–75. doi: 10.1006/dbio.2002.0685. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Jun S, Desplan C. Cooperative interactions between paired domain and homeodomain. Development. 1996 Sep;122(9):2639–50. doi: 10.1242/dev.122.9.2639. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Buckingham ME. The anterior heart-forming field: voyage to the arterial pole of the heart. Trends Genet. 2002 Apr;18(4):210–6. doi: 10.1016/s0168-9525(02)02642-2. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Hunt P, Wallis K, Thorogood P. Abnormal patterning of the aortic arch arteries does not evoke cardiac malformations. Dev Dyn. 1997;208(1):34–47. doi: 10.1002/(SICI)1097-0177(199701)208:1<34::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kirby ML. Contribution of Neural Crest to Heart and Vessel Morphology. In: Harvey RP, Rosenthal N, editors. Heart Development. London: Academic Press; 1999. pp. 179–193. [Google Scholar]
- Koushik SV, Wang J, Rogers R, Moskofidis D, Lambert L, Creazzo T, Conway SJ. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 2001 doi: 10.1096/fj.00-069fje. March 12, 2001. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Oda H, Maemura K, Nagai R, Ishikawa T, Yazaki Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J Clin Invest. 1995;96(1):293–300. doi: 10.1172/JCI118033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, Maas RL, Maxson RE., Jr Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129(2):527–3. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- LaBonne C, Bronner-Fraser M. Molecular mechanisms of neural crest formation. Annu Rev Cell Dev Biol. 1999;15:81–112. doi: 10.1146/annurev.cellbio.15.1.81. [DOI] [PubMed] [Google Scholar]
- Lagutina I, Conway SJ, Sublett J, Grosveld G. PAX3-FKHR knock-in mice show developmental aberrations but do not develop tumors. Molecular and Cellular Biology. 2002 doi: 10.1128/MCB.22.20.7204-7216.2002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol. 1975;34(1):125–54. [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126(11):2495–503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Li L, Heller-Harrison R, Czech M, Olson EN. Cyclic AMP-dependent protein kinase inhibits the activity of myogenic helix-loop-helix proteins. Mol Cell Biol. 1992a;12:4478–4485. doi: 10.1128/mcb.12.10.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhou J, James G, Heller-Harrison R, Czech MP, Olson EN. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell. 1992b;71:1181–1194. doi: 10.1016/s0092-8674(05)80066-2. [DOI] [PubMed] [Google Scholar]
- Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–74. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Baldini A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Hum Mol Genet. 2001;10(9):997–1002. doi: 10.1093/hmg/10.9.997. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128(11):2039–48. doi: 10.1242/dev.128.11.2039. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002 Nov;129(21):5081–91. doi: 10.1242/dev.129.21.5081. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Pla P, Larue L, Gruss P. Pax3 acts cell autonomously in the neural tube and somites by controlling cell surface properties. Development. 2001;128(11):1995–2005. doi: 10.1242/dev.128.11.1995. [DOI] [PubMed] [Google Scholar]
- Markus M, Benezra R. Two isoforms of protein disulfide isomerase alter the dimerization status of E2A proteins by a redox mechanism. Journal of Biological Chemistry. 1999;274:1040–1049. doi: 10.1074/jbc.274.2.1040. [DOI] [PubMed] [Google Scholar]
- Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet. 2000;97(4):280–8. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-Loop-Helix Proteins: Regulators of Transcription in Eucaryotic Organisms. Molec Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Bourges E, Caron F, Piussan C. Waardenburg's syndrome and severe cyanotic cardiopathy. Arch Fr Pediatr. 1990;47:657–659. [PubMed] [Google Scholar]
- Mercola M, Levin M. Left-right asymmetry determination in vertebrates. Annual Review of Cell and Developmental Biology. 2001;17:779–805. doi: 10.1146/annurev.cellbio.17.1.779. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–29. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Miller CT, Schilling TF, Lee K, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- Miskiewicz P, Morrissey D, Lan Y, Raj L, Kessler S, Fujioka M, Goto T, Weir M. Both the paired domain and homeodomain are required for in vivo function of Drosophila Paired. Development. 1996 Sep;122(9):2709–18. doi: 10.1242/dev.122.9.2709. [DOI] [PubMed] [Google Scholar]
- Momma K, Matsuoka R, Takao A. Aortic arch anomalies associated with chromosome 22q11 deletion (CATCH 22) Pediatr Cardiol. 1999;20(2):97–102. doi: 10.1007/s002469900414. [DOI] [PubMed] [Google Scholar]
- Noden DM. Embryonic origins and assembly of blood vessels. Am Rev Respir Dis. 1989;140:1097–103. doi: 10.1164/ajrccm/140.4.1097. [DOI] [PubMed] [Google Scholar]
- Olson EN. Regulation of muscle transcription by the MyoD family. The heart of the matter. Circ Res. 1993;72:1–6. doi: 10.1161/01.res.72.1.1. [DOI] [PubMed] [Google Scholar]
- Olson EN, Klein WH. bHLH factors in muscle development: dead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Olson EN, Srivastava D. Molecular pathways controlling heart development. Science. 1996;272(5262):671–6. doi: 10.1126/science.272.5262.671. [DOI] [PubMed] [Google Scholar]
- Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002 Jun 4;12(11):938–43. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- Riley P, Anson-Cartwright L, Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet. 1998;18:271–275. doi: 10.1038/ng0398-271. [DOI] [PubMed] [Google Scholar]
- Stottmann Rolf W, Murim Choi, Mishina Yuji, Meyers Erik N, Klingensmith John. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004 doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satokata I, Ma L, Ohshima H, Bei M, Woo I, Nishizawa K, Maeda T, Takano Y, Uchiyama M, Heaney S, Peters H, Tang Z, Maxson R, Maas R. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet. 2000;24(4):391–5. doi: 10.1038/74231. [DOI] [PubMed] [Google Scholar]
- Schafer K, Neuhaus P, Kruse J, Braun T. The homeobox gene Lbx1 specifies a subpopulation of cardiac neural crest necessary for normal heart development. Circ Res. 2003 Jan 10;92(1):73–80. doi: 10.1161/01.res.0000050587.76563.a5. [DOI] [PubMed] [Google Scholar]
- Schubert FR, Tremblay P, Mansouri A, Faisst AM, Kammandel B, Lumsden A, Gruss P, Dietrich S. Early mesodermal phenotypes in splotch suggest a role for Pax3 in the formation of epithelial somites. Dev Dyn. 2001;222(3):506–21. doi: 10.1002/dvdy.1211. [DOI] [PubMed] [Google Scholar]
- Serbedzija GN, McMahon AP. Analysis of neural crest cell migration in Splotch mice using a neural crest-specific LacZ reporter. Dev Biol. 1997;185(2):139–47. doi: 10.1006/dbio.1997.8551. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nature Genetics. 1997;16:154–160. doi: 10.1038/ng0697-154. [DOI] [PubMed] [Google Scholar]
- Srivastava D. Genetic assembly of the heart: implications for congenital heart disease. Annu Rev Physiol. 2001;63:451–69. doi: 10.1146/annurev.physiol.63.1.451. [DOI] [PubMed] [Google Scholar]
- Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Gewillig M, Molin DG, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Plaisance S, Vlietinck R, Emanuel B, Gittenberger-de Groot AC, Scambler P, Morrow B, Driscol DA, Moons L, Esguerra CV, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P. VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med. 2003 Feb;9(2):173–82. doi: 10.1038/nm819. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and DiGeorge anomaly: critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am J Med Genet Suppl. 1986;2:97–112. doi: 10.1002/ajmg.1320250614. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Liu XZ, Brady A, Donnai D, Krajewska-Walasek M, Murday V, Norman A, Obersztyn E, Reardon W, et al. The mutational spectrum in Waardenburg syndrome. Hum Mol Genet. 1995;4(11):2131–7. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- Thomas T, Kurihara H, Yamagishi H, Kurihara Y, Yazaki Y, Olson EN, Srivastava D. A signaling cascade involving endothelin-1, dHAND and msx1 regulates development of neural-crest-derived branchial arch mesenchyme. Development. 1998;125:3005–3014. doi: 10.1242/dev.125.16.3005. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, Wessels A. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–490. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;15(118):915–22. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Lo CW, Kirby ML. Connexin 43 expression reflects neural crest patterns during cardiovascular development. Dev Biol. 1999a;208(2):307–23. doi: 10.1006/dbio.1999.9219. [DOI] [PubMed] [Google Scholar]
- Waldo K, Zdanowicz M, Burch J, Kumiski DH, Stadt HA, Godt RE, Creazzo TL, Kirby ML. A novel role for cardiac neural crest in heart development. J Clin Invest. 1999b;103(11):1499–507. doi: 10.1172/JCI6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WP, Widelitz RB, Jiang TX, Chuong CM. Msx-2 and the regulation of organ size: epidermal thickness and hair length. J Investig Dermatol Symp Proc. 1999;4(3):278–81. doi: 10.1038/sj.jidsp.5640229. [DOI] [PubMed] [Google Scholar]
- Wawersik S, Maas RL. Vertebrate eye development as modeled in Drosophila. Hum Mol Genet. 2000 Apr 12;9(6):917–25. doi: 10.1093/hmg/9.6.917. [DOI] [PubMed] [Google Scholar]
- Wendling O, Dennefeld C, Chambon P, Mark M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development. 2000;127(8):1553–62. doi: 10.1242/dev.127.8.1553. [DOI] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9(17):2105–16. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Winnier G, Kume T, Deng K, Rogers R, Bundy J, Raines C, Hogan BLM, Conway SJ. Roles for the winged helix transcription factors MF1 and MFH1 in cardiovascular development revealed by non-allelic non-complementation of null alleles. Developmental Biolog. 1999;216:16–27. doi: 10.1006/dbio.1999.9382. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Hammer RE, Richardson JA, Williams SC, Clouthier DE, Yanagisawa M. Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aortic arch patterning in mice. J Clin Invest. 1998;102(1):22–33. doi: 10.1172/JCI2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa H, Clouthier DE, Richardson JA, Charite J, Olson EN. Targeted deletion of a branchial arch-specific enhancer reveals a role of dHAND in craniofacial development. Development. 2003;130:1069–1078. doi: 10.1242/dev.00337. [DOI] [PubMed] [Google Scholar]
- Ziman MR, Kay PH. A conserved TN8TCCT motif in the octapeptide-encoding region of Pax genes which has the potential to direct cytosine methylation. Gene. 1998 Nov 26;223(12):303–8. doi: 10.1016/s0378-1119(98)00162-0. [DOI] [PubMed] [Google Scholar]