

Abstract
5,6-Dihydro-5-fluorouracil (5-DHFU) is a metabolite of the chemotherapy drug 5-fluorouracil (5-FU) of importance for biological studies. 5-DHFU has been prepared by enzymatic reduction of 5-FU and in very low yield by hydrogenation of 5-FU; however, a practical chemical synthesis is not available. Facile racemic syntheses of 5-DHFU from 5-FU or uracil using p-methoxybenzyl protecting groups followed by L-Selectride reduction are reported.
5-Fluorouracil (5-FU) was discovered in 1957, and today it is a widely prescribed antineoplastic drug, most commonly used in the treatment of breast and colorectal cancer.1 There are two mechanisms by which 5-FU kills cancer cells. It is known to bind to the nucleotide binding site of thymidylate synthase, thereby inhibiting deoxythymidine monophosphate synthesis, which affects the levels of other deoxynucleotides and interrupts DNA synthesis and repair. 5-FU can also be misincorporated into DNA and RNA in place of uracil, which leads to mutations and induction of cell death.2,3
As with many chemotherapeutic treatments, a major problem associated with the use of 5-FU is its lack of selectivity between cancerous and normal cells. This in turn causes a number of side effects, some of which can be fatal. In the case of 5-FU, the side effects are attributed to a lack of the enzyme dihydropyrimidine dehydrogenase (DPD), which metabolizes 5-FU into R-5,6-dihydro-5-fluorouracil (5-DHFU), as seen in Figure 1.
Figure 1.
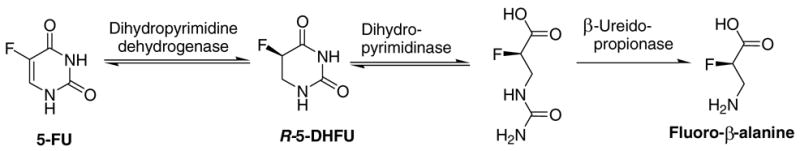
The DPD step is rate limiting and is followed by two subsequent enzymatic conversions to low-toxicity fluoro-β-alanine.3,4 In patients lacking the DPD enzyme, 5-FU accumulates to levels that begin to destroy normal cells.
5-DHFU is about 100-fold less toxic to human cells than 5-FU, but it is not as effective in killing cancer cells.3 For this reason, it may be advantageous to deliver 5-DHFU as a prodrug and design a way to convert it to 5-FU in vivo, particularly if this can be done locally, in a controlled fashion. 5-DHFU has been prepared in 50% yield by enzymatic reduction using bovine DPD,5 a process with only limited convenience and scalability, and chemically by hydrogenation of 5-FU in 6.5% yield.6 To further investigate the biological properties of 5-DHFU, we developed improved chemical syntheses starting with 5-FU and uracil by which 5-DHFU can be produced efficiently and in larger quantities.
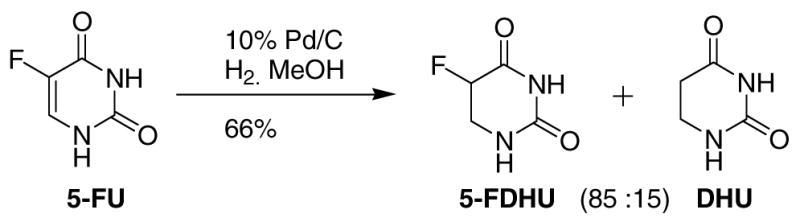
The most direct synthetic route to 5-DHFU from 5-FU is hydrogenation of the C=C bond (Scheme 1). This was attempted using 10 mol% palladium on carbon (Pd/C) as catalyst in methanol under an atmosphere of hydrogen at room temperature for 36 hours. 1H NMR of the product indicated that it was an 85:15 mixture of 5-DHFU and 5,6-dihydrouracil (DHU), the latter being formed by hydrogenolysis of the C-F bond. These results are similar to those reported previously on the hydrogenation of 5-FU.5 Hydrogenolysis of vinylic and aliphatic C-F bonds is a common problem under these conditions.7,8 Unfortunately, recrystallization of the product mixture failed to remove the DHU, and flash chromatography was not a practical option: Both 5-DHFU and DHU are very polar and insoluble in most organic solvents, with the exception of DMSO and DMF; furthermore, neither compound is short wave UV active, making it difficult to visualize them on TLC. 5-FU and uracil are, however, UV active. A large number of TLC staining techniques were examined, but none of them proved to be effective. This combination of factors made separation of this product mixture by flash chromatography impractical.
Scheme 1.
To determine whether hydrogenolysis of the C -F bond occurred in concert with or after the reduction of the C=C bond, the kinetics of the reduction reactions were studied by 1H NMR. If cleavage occurred as the second step, we surmised that the reaction could be stopped at low conversion and the 5 -DHFU separated from the 5 -FU. Two hydrogenation reactions were carried out, one at room temperature and one at 0 °C, to determine whether temperature had any effect on the product ratio. Aliquots were taken from each reaction every two hours, filtered to remove the Pd catalyst and concentrated. The samples were redissolved in DMSO-d6, and NMR spectra were obtained. Integration of the peaks in the NMR spectrum was used to monitor consumption of 5-FU and formation of 5-DHFU and DHU.
A plot of the percent composition of each component versus time in the 25 °C reaction was generated from the 1H NMR data (Figure 2). It is clear that C-F hydrogenolysis occurs early in the course of the reaction, beginning around 25% conversion of 5-FU. As expected, reducing the temperature to 0 °C slowed the reaction, but it did not delay the formation of DHU (not shown). In both cases, a small amount of uracil was observed in the NMR spectra at low conversion, further indicating that C-F cleavage occurs early in the reaction. Because the DHU formed so early, the reaction would need to be stopped at 10–15% conversion to obtain 5-DHFU essentially without DHU present, which is not a practical option.
Figure 2.

Plot of the data obtained from 1H NMR analysis of the reaction composition in the kinetic study of 5-FU hydrogenation at 25 °C. Solid = 5-FU, Dashed = 5-DHFU, Dotted = DHU.
Before abandoning the hydrogenation approach, some other catalysts and conditions were investigated. Poisoning the Pd/C catalyst with Et3N was attempted; this slowed the reaction considerably but did not alter the 5-DHFU:DHU ratio. Lower Pd/C catalyst loading (5% vs 10%), different solvents (H2O, CH3OH/H2O) and increased temperature also had no beneficial effect. Hydrogenation using the homogenous Wilkinson’s catalyst (RhCl[PPh3]3) was unsuccessful, with no conversion of 5-FU occurring. PtO2 was also employed as a hydrogenation catalyst; it did not reduce the C=C bond, but it did cleave the C-F bond, giving uracil.
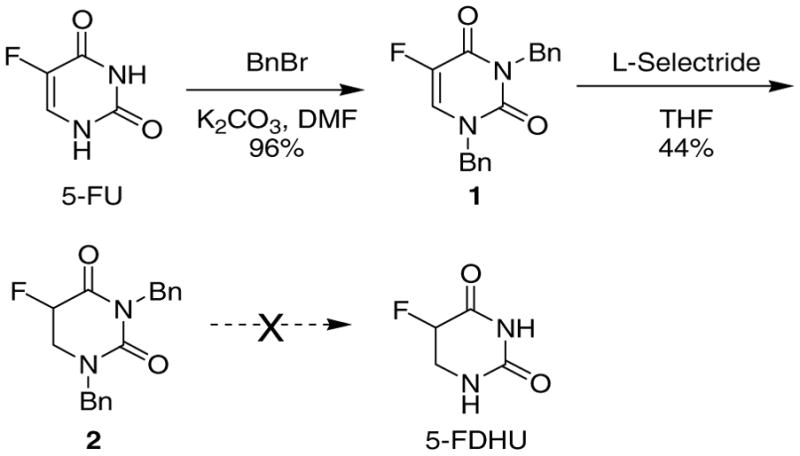
After several failed attempts at preparing 5-DHFU by hydrogenation, a thorough search of the literature yielded an article describing the reduction of the C=C bond in uracil derivatives using lithium tri-sec-butylborohydride (L-Selectride) to give the corresponding DHU derivative.9 We attempted this procedure using 5-FU, as shown in Scheme 2. In the first step, the amide nitrogens on 5-FU were protected with benzyl (Bn) groups in excellent yield, to lock 5-FU in the “ene-dione” tautomeric form, increase its solubility and render it UV visible and prevent quenching of the L-Selectride during reduction.9 In the subsequent step, benzyl-protected 1 was reduced using a 1 M solution of L-Selectride in THF (from Sigma-Aldrich) in moderate yield, and the reaction product 2 was easily purified by flash column chromatography. However, removal of the benzyl groups in 2 by hydrogenation,10 dissolving metal reduction,11 or treatment with Lewis acids12 to afford 5-DHFU proved to be problematic.
Scheme 2.
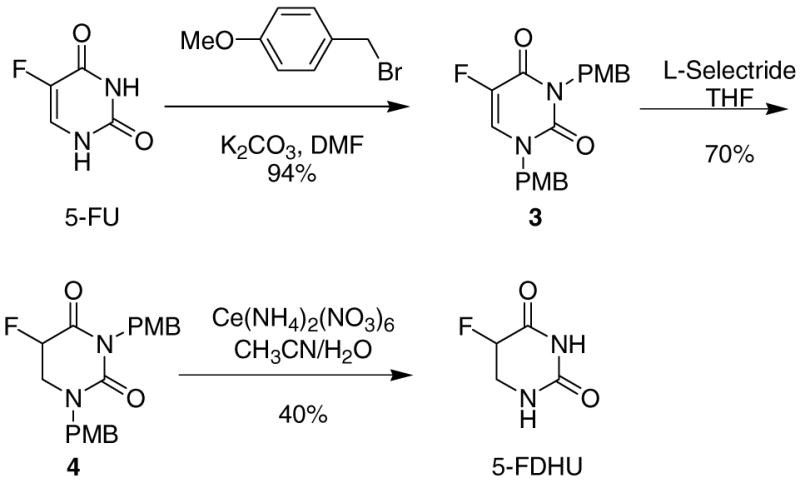
Due to the difficulties we encountered in trying to remove the N-benzyl group, we sought an alternate protecting group. There are many reports in the literature of methoxymethyl (MOM) as an N-protecting group for amides; though base stable, the MOM group is quite labile in acid and is usually cleaved with dilute HCl or a Lewis acid (AlCl3, BBr3).13 Both amide nitrogens of 5-FU were MOM-protected in 94% yield.14 Reduction of the MOM-protected 5-FU using L-Selectride occurred in good yield. Removal of the MOM protecting groups was attempted using a variety of conditions,10 with little success.
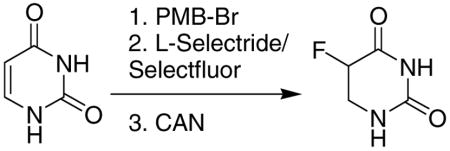
After further unsuccessful attempts at either installing or removing trimethylsilyl, acetyl, benzoyl, tosyl and nosyl as protecting groups, p-methoxybenzyl (PMB) was finally found to be a suitable option. 5-FU could be easily protected with this group in a similar fashion as in the preparation of the simple bis-benzyl protected compound 1 (Scheme 2). As with 2, L-Selectride reduction of 3 gave reduced product 4 cleanly, in 70% yield. Deprotection of 4 using neat refluxing TFA15 or AlCl3 in anisole16 was unsuccessful. Ceric ammonium nitrate (CAN) in 3:1 CH3CN and water,10 however, removed both PMB groups to afford 5-DHFU, although isolation of this polar product from the salt byproducts required continuous extraction, performed according to the following isolation protocol: The reaction solvent was evaporated from the crude product, and the resulting solid was redissolved in a small volume (~20 mL) of water and then washed briefly with CHCl3 to remove the organic byproducts. The aqueous layer containing 5-FDHU was then transferred to a continuous extractor and extracted with EtOAc for 24–36 hours. The extraction solvent was removed by evaporation, and the resulting solid was recrystallized from ethanol to give very pure 5-DHFU.
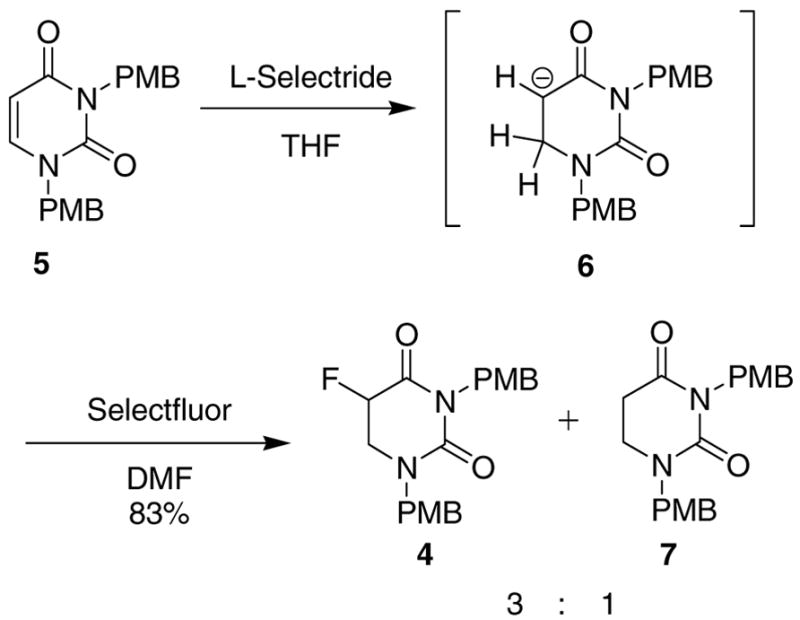
An alternative method was developed to prepare 5-DHFU from uracil instead of 5-FU. 5-FU is currently over 10 times more expensive than uracil, so using uracil as a starting material has significant cost advantages. Uracil was N-alkylated with 4-methoxybenzyl bromide in a similar manner to 3. The L-Selectride reduction goes through intermediate 6 (Scheme 4), and because Kundu et al. were able to alkylate this position, we wondered whether it could be fluorinated electrophilically. L-Selectride reduction of 5 followed by treatment with Selectfluor (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) in anhydrous DMF (instead of acidic workup) gave the desired product (4) in 58% yield, as well as the protio compound 7, in 25% yield; formation of the latter product was presumed due to the presence of water in the DMF. The products were easily separated using silica gel flash chromatography (1:1 EtOAc/hexanes).
Scheme 4.
This work demonstrates the first practical chemical syntheses of 5-DHFU, and it opens possibilities for using this material in various biological applications.
Experimental
Hydrogenation of 5-Fluorouracil (5-FU)
5-Fluorouracil (790 mg, 6.1 mmol) was dissolved in methanol (100 mL) after vigorous stirring for 1 hr. Pd/C (10 wt %, 646 mg, 0.61 mmol) was added, and the mixture was stirred under a hydrogen atmosphere for 36 h at room temperature. The reaction mixture was diluted with methanol, filtered through Celite and then concentrated. The product was recrystallized from water, resulting in a white solid (530 mg, 66%) which was an inseparable 85:15 mixture of 5,6-dihydro-5-fluorouracil and 5,6-dihydrouracil (as determined by 1H NMR integration).
1,3-Bis-(4-methoxybenzyl)-5-fluorouracil (3)
5-Fluorouracil (260 mg, 2 mmol) and K2CO3 (663 mg, 4.8 mmol) were added to an oven-dried 3-neck flask under argon. Anhydrous DMF (5 mL) was added, and the mixture was stirred for 1 h, resulting in a thick gel. DMF (2 mL) and 4-methoxybenzyl bromide (0.87 mL, 6 mmol) were added. and the reaction was stirred for another 24 h until completion was ascertained by TLC analysis (2:1 hexanes/EtOAc). The reaction mixture was diluted with EtOAc and washed with water and brine, dried over MgSO4, filtered and concentrated. Purification by silica gel column chromatography (1:1 EtOAc/Hexanes) afforded the product as a white solid (693 mg, 94%). 1H NMR (CDCl3, 500 MHz) δ: 7.50 (m, 2H), 7.22 (m, 2H), 7.14 (d, J = 5.36 Hz, 1H), 6.89 (m, 2H), 6.85 (m, 2H), 5.10 (s, 2H), 4.83 (s, 2H), 3.81 (s, 3H), 3.79 (s, 3H). 13C NMR (CDCl3, 125 MHz) δ: 159.6, 159.0, 157.0 (d, J = 25.1 Hz), 150.0, 140.8 (d, J = 235.67 Hz), 130.6, 129.6, 128.1, 126.1, 125.6, 125.3, 114.3, 113.4, 55.0 (d, J = 14.73 Hz), 51.5, 44.3. HRMS: Calc’d for C20H19FN2O4 [M+]: 371.1407. Found: 371.1423.
1,3-Bis-(4-methoxybenzyl)-uracil (5)
Uracil (930 mg, 8.29 mmol) and K2CO3 (2.75 g, 19.89 mmol) were added to an oven-dried 3-neck flask under argon. Anhydrous DMF (35 mL) was added, and the mixture was stirred for 18 h, resulting in a thick gel. 4-Methoxybenzyl bromide (3.6 mL [5 g], 24.87 mmol) was added, and the reaction was stirred for another 5 days. The reaction mixture was concentrated and redissolved in water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over MgSO4, filtered and concentrated. Purification by silica gel column chromatography (2:1 EtOAc/hexanes) afforded the product (5) as a white solid (1.066 g, 36%). 1H NMR (CDCl3, 500 MHz) δ: 7.47 (m, 2H), 7.21 (m, 2H), 7.08 (d, J = 7.93 Hz), 6.89 (m, 2H), 6.83 (m, 2H), 5.70 (d, J = 7.93 Hz), 5.07 (s, 2H), 4.82 (s, 2H), 3.79 (s, 3H), 3.77 (s, 3H). 13C NMR (CDCl3, 125 MHz) δ: 162.8, 159.6, 158.9, 151.6, 141.5, 130.5, 129.5, 129.0, 127.0, 114.3, 113.5, 101.8, 55.2, 55.0, 51.6, 43.6. HRMS: Calc’d for C20H20N2O4 [M+]: 353.1501. Found: 353.1501
1,3-Bis-(4-methoxybenzyl)-5,6-dihydro-5-fluorouracil (4)
Method A
(By hydride reduction of bis-PMB-5-FU): A flask containing a stir bar and 1,3-(4-methoxybenzyl)-5-fluorouracil (3, 2.05 g, 5.53 mmol) was evacuated and purged with argon three times. Anhydrous THF (25 mL) was added, and the reaction was cooled to −78 °C. L-Selectride (1 M in THF, 6.08 mL, 6.08 mmol) was added via syringe and stirred for 90 min at −78 °C. Saturated NH4Cl was added, and then the mixture was warmed to room temperature and stirred for 30 min. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with water and brine, dried over MgSO4, filtered and concentrated. Purification by silica gel column chromatography (1:1 EtOAc/hexanes) afforded the product (4) as a clear oil (1.44 g, 70%).
Method B
(By hydride reduction-fluorination of 5): A flask containing a stir bar and 1,3-(4-methoxybenzyl)-uracil (5, 1.26 g, 3.56 mmol) was evacuated and purged with argon three times. Anhydrous THF (15 mL) was added, and the reaction was cooled to −78 °C. L-Selectride (1 M in THF, 3.93 mL, 3.93 mmol) was added via syringe and stirred for 2 h at −78 °C. A solution of SelectFluor (1.39 g, 3.93 mmol) in anhydrous DMF (5 mL) was added, and the reaction was slowly warmed to room temperature and stirred overnight. The solvent was evaporated and redissolved in EtOAc, washed with water and brine, dried over MgSO4, filtered and concentrated. Purification by silica gel column chromatography (1:1 EtOAc/hexanes) afforded the product (4) as a clear oil (766 mg, 58%). N,N-Bis-(4-methoxybenzyl)-5,6-dihydrouracil (8) was also isolated as a clear oil (325 mg, 25%), because protonation competes with fluorination. 1H NMR (CDCl3, 500 MHz) δ: 7.40 (m, 2H), 7.19 (m, 2H), 6.85 (m, 4H), 4.96 (d, J = 4.07 Hz, 2H), 4.94 (m, 1H), 4.58 (dd, J = 37.3, 14.5 Hz, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 3.39 (m, 2H). 13C NMR (CDCl3, 125 MHz) δ: 165.1 (d, J = 20.5 Hz), 159.5, 159.0, 152.3, 130.4, 129.5, 129.2, 127.5, 114.3, 113.7, 82.3 (d, J = 185.5 Hz), 55.2, 51.0, 44.7 (d, J = 26.2 Hz), 43.9. HRMS: Calc’d for C20H21FN2O4 [M+]: 373.1564. Found: 373.1576.
5,6-Dihydro-5-fluorouracil (5-DHFU)
1,3-Bis-(4-methoxybenzyl)--5,6-dihydro-5-fluorouracil (4, 1.26 g, 3.39 mmol) was dissolved in CH3CN (20 mL). Ceric ammonium nitrate (7.43 g, 13.55 mmol) was added followed by water (7 mL), and the reaction was stirred for 3 h at room temperature until completion was ascertained by TLC analysis (2:1 EtOAc/hexanes). The solvent was evaporated, and the resulting solid was redissolved in water (20 mL) and washed with CHCl3 (3 × 50 mL). The aqueous layer was continuously extracted with EtOAc for 36 h using a liquid-liquid continuous extractor. The EtOAc was evaporated, and the product was recrystallized from ethanol and isolated as a white solid (140 mg, 31%), which decomposed at 260 °C (Lit. 241–243 °C decomposed)6.1H NMR (DMF-d7, 500 MHz) δ: 10.49 (br. S, 1H), 7.71 (br. S, 1H), 5.26 (ddd, J = 47.2, 7.7, 5.1 Hz, 1H), 3.79 (dddd, J = 20.9, 13.1, 5.0, 3.1 Hz), 3.60 (dddd, J = 12.8, 12.7, 7.7, 2.4 Hz, 1H). 13C NMR (DMF-d7, 125 MHz) δ: 168.2 (d, J = 20.2 Hz), 154.0, 84.5 (d, J = 178.7 Hz), 42.1 (d, J = 25.2 Hz). HRMS: Calc’d for C4H5FN2O2[M + H+]: 133.0413. Found: 133.0420.
Supplementary Material
Experimental details, 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.
Acknowledgments
We thank Professor Michael Strano at the University of Illinois at Urbana-Champaign for presenting this synthetic challenge to us. This work was supported by a grant from the National Institutes of Health. ESI mass spectra were recorded on a spectrometer purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 07141).
References
- 1.Bocci G, Danesi R, Allegrini G, Innocenti F, Di Paolo A, Falcone A, Conte PF, Del Tacca M. Eur J Clin Pharmacol. 2002;58:593–595. doi: 10.1007/s00228-002-0534-6. [DOI] [PubMed] [Google Scholar]
- 2.Longley DB, Harkin PD, Johnston PG. Nature Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 3.Fischel J, Formento P, Ciccolini J, Etienne-Grimaldi M, Milano G. Anti-Cancer Drugs. 2004;15:969–974. doi: 10.1097/00001813-200411000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Van Kuilenburg ABP, Meinsma R, van Gennip AH. Nucleosides, Nucleotides and Nucleic Acids. 2004;23:1371–1375. doi: 10.1081/NCN-200027624. [DOI] [PubMed] [Google Scholar]
- 5.Porter DJT, Harrington JA, Almond MR, Lowen GT, Spector T. Biochemical Pharmacology. 1994;48:775–779. doi: 10.1016/0006-2952(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 6.Duschinsky R, Pleven E, Heidelberger C. J Am Chem Soc. 1957;79:4559–4560. [Google Scholar]
- 7.Allmendinger T, Dandois C, Walliser B. Tetrahedron Lett. 1991;32:2735–2736. [Google Scholar]
- 8.Engman M, Diesen JS, Paptchikhine A, Andersson PG. J Am Chem Soc. 2007;129:4536–4537. doi: 10.1021/ja0686763. [DOI] [PubMed] [Google Scholar]
- 9.Kundu NG, Sikdar S, Hertzberg RP, Schmitz SA, Khatri SG. J Chem Soc Perkin Trans I. 1985:1295–1300. [Google Scholar]
- 10.Wuts PGM, Greene TW. Protective Groups in Organic Synthesis. 3. Wiley Interscience; 1999. [Google Scholar]
- 11.Magdolen P, Vasella A. Helv Chim Acta. 2005;88:2454–2469. [Google Scholar]
- 12.Coelho A, Sotelo E, Novoa H, Peeters OM, Blaton N, Ravina E. Tetrahedron. 2004;60:12177–12189. [Google Scholar]
- 13.Wu F, Buhendwa MG, Weaver DF. J Org Chem. 2004;69:9307–9309. doi: 10.1021/jo0485076. [DOI] [PubMed] [Google Scholar]
- 14.Heintzelman GR, Fang W, Keen SP, Wallace GA, Weinreb SM. J Am Chem Soc. 2002;124:3939–3945. doi: 10.1021/ja020032h. [DOI] [PubMed] [Google Scholar]
- 15.Brooke GM, Mohammed S, Whiting MC. Chem Commun. 1997:1511–1512. [Google Scholar]
- 16.Akiyama T, Takesue Y, Kumegawa M, Nishimoto H, Ozaki S. Bull Chem Soc Jpn. 1991;64:2266–2269. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.